Andersen-Tawil syndrome
Andersen Tawil syndrome (ATS), also known as long QT syndrome type 7 (LQT7), is a rare genetic disease characterized by ventricular arrhythmias, periodic paralysis, and abnormal physiological morphology. Klein et al. first mentioned the syndrome in 1963. They described a special form of periodic paralysis with ventricular premature beats. In 1971 Ellen Andersen first described the three groups of characteristics caused by this disease. However, it was not until 1994 that Tawil and colleagues described multiple patients that the syndrome was correctly described, so the disease was called Anderson syndrome. In 2003, due to the important contribution of Tavel to the clinical research of the syndrome, it was later changed to Anderson Tavel syndrome.
Pathogenesis of ATS
Anderson's syndrome is caused by genetic disease mutations in the KCNJ2 gene in most cases. The disease is usually inherited from parents through autosomal dominant inheritance, but may also occur due to new genetic mutations in the sick. The manifestations of ATS-related symptoms are diverse, and not all patients with ATS exhibit the same symptoms. So far, only mutations in the KCNJ2 gene have been found to cause ATS. This gene encodes Kir2.1, an inwardly rectifying potassium channel protein. Clinically, ATS patients with this gene defect are usually designated as ATS1 (ATS1), and ATS caused by unknown mutations are considered to be ATS2 (ATS2).
Pathophysiology of ATS
The inward rectifier potassium channel protein Kir2.1 is encoded by the KCNJ2 gene, which is mainly expressed in the heart and skeletal muscle tissue. In the human heart, Kir2.1 is abundantly expressed in the ventricle, but less in the atrium. Structural studies found that Kir2.1 protein is composed of a protein with two transmembrane helices and a pore-forming loop. Among them, the amino terminal and the carboxy terminal are located in the cell, and there is a sliding helix near the N terminal. The study found that the subunits of Kir2.x can form functional channels of tetramers through heteromerization or homomerization. The channel thus formed forms the basis of the inward rectified potassium current IK1. As we all know, muscle contraction of the heart and skeletal muscle cells is achieved by changes in the action potential caused by the ion current. When heart and skeletal muscle cells are stimulated, the cell membrane reaches the action potential from the resting membrane potential. After the heart and skeletal muscle cells relax, the inside of the cell membrane has less positively charged ions than the outside, which is the polarized membrane. The main ion current responsible for maintaining this polarity is IK1, and the decrease of this current causes the polarity at rest to become smaller or the static membrane potential to be depolarized.When these cells contract, positively charged ions such as sodium and calcium enter the cell through ion channels, depolarizing or reversing the polarity.
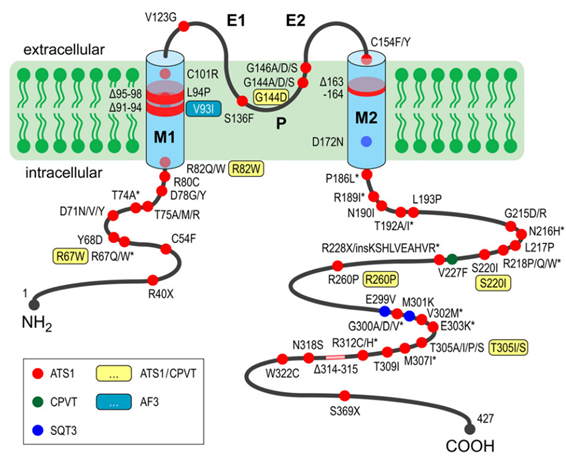
Figure 1. Mutations in KCNJ2 associated with cardiac arrhythmia syndromes. (Nguyen HL, et al. 2013)
After contraction occurs, the cell restores its polarity (or repolarizes) by allowing positively charged ions (such as potassium) to leave the cell and restore the membrane to its relaxed polarized state. It has been found in Andersen-Tawil patients that KCNJ2 gene mutations reduce potassium flow and slow the rate of repolarization. This effect shows a prolonged action potential in a single cardiomyocyte, while an extended QT interval is detected on the heart detection. Because IK1 plays an important role in maintaining a stable resting membrane potential in the excitable cells and cardiac action potential during the repolarization period, this may explain why IK1 channel defects can cause prolonged QT interval and arrhythmia in patients with ATS. It is worth noting that the researchers also found that abnormal U-waves appearing in the electrocardiogram of ATS patients were negatively correlated with IK1, that is, the absence of IK1 in ATS would cause abnormal U-wave ECG signals in ATS patients. In addition to the heart, defects in the Kir2.1 channel in skeletal muscle tissue reduce membrane excitability, which in turn causes potassium ion imbalance, which ultimately leads to muscle weakening and periodic paralysis.
References
- Nguyen HL, et al. Andersen-Tawil syndrome: Clinical and molecular aspects. International Journal of Cardiology. 2013, 170(1): 1-16.
- Wit, Andrew L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing and Clinical Electrophysiology: PACE. 2018, 41 (8): 883-896.
Inquiry
