Autosomal Dominant Polycystic Kidney Disease
Polycystic kidney disease (PKD) is a common autosomal inherited disease, which is single-gene inherited. It is genetically divided into dominant and recessive categories. Both types cause bilateral kidney disease. Among them, autosomal dominant polycystic kidney disease (ADP KD) is the most common, mainly characterized by the continuous formation and expansion of fluid-filled vesicles derived from renal tubular epithelial cells, and typical cases will develop into end-stage kidneys in middle age stages of disease. ADPKD is more common than adulthood, with high incidence and poor prognosis. At present, there is no effective treatment in clinic. Therefore, since the first pathogenic gene of ADPKD was discovered, researchers have done a lot of research and exploration on its pathogenesis and treatment methods.
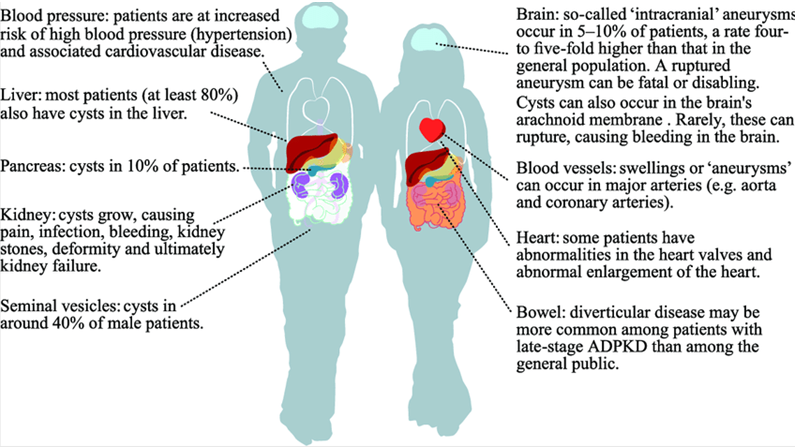
Figure 1. Parts of the body that may be affected by autosomal dominant polycystic kidney disease. (Tess Harris, 2018)
ADPKD is the most common type of polycystic kidney, and the progressive formation and increased cysts lead to the progressive enlargement of the kidneys on both sides is its main feature. ADPKD is accompanied by multiple extrarenal manifestations, often damaging the liver, spleen, and blood vessels. The age of onset of patients is generally older, mostly after the age of 40, there are more low back pain, upper abdominal pain, hypertension, hematuria and abdominal masses, etc. The appearance of these clinical manifestations is often a sign that the renal parenchyma has been severely damaged, and more seriously These damages will continue to increase. Although most patients develop the disease after middle age, there is currently a lot of evidence that ADPKD can be diagnosed as early as the fetal period, and the disease is more serious than the adult. Selistre et al's study of 52 ADPKD patients with childhood onset confirmed the exact damage to the kidney site, and 77% of them developed hypertension, proteinuria, and renal function damage. Therefore, when diagnosing ADPKD, it should not only be limited to middle-aged patients, but more attention should be paid to early detection of early-onset patients, and timely intervention to delay disease progression and improve their quality of life.
ADPKD Molecular Mechanism
85% of ADPKD patients are from PKD1 gene mutation, the gene is located on the short arm of chromosome 16, the product is polycystin 1 (PC1); about 15% are from the gene PKD2 mutation, located on the long arm of chromosome 4, the product is polycystin 2 (PC2). In the past, it was believed that some ADPKD originated from a PKD3 gene mutation, but Paul et al. found that this mutation is equivalent to the PKD1 and PKD2 mutations, and therefore denied the existence of PKD3. PC1 is widely distributed, including kidney, liver, pancreas, heart and small intestine. It participates in various signal transduction as a membrane receptor. In the kidney, polycystin 1 mutations will produce various abnormalities: the signal that regulates cell proliferation will be affected, and the consequence is the continuous proliferation of cystic epithelial cells; if PC1 loss occurs on the cell surface, it will cause the adhesion between the cells breaks, eventually changing the polarity of the tubules, and the PC1 fusion protein appears, which promotes the continuous accumulation of cyst fluid and the continuous expansion of the cyst cavity. PC2 is a non-selective cation channel that allows calcium ion permeability and participates in the positioning of PC1. It is mainly expressed in the endoplasmic reticulum, the outer side of the cell membrane, and the primary cilia. Diseases caused by PKD2 mutations are relatively lighter than PKD1 mutations. Cornec-Le et al. found that among the PKD1 mutations, truncation mutations are more serious than missense mutations in the kidney phenotype. These new discoveries in molecular genetics make future research on ADPKD more targeted and make it easier to achieve key breakthroughs.
References
- Torres VE, et al.; Autosomal dominant polycystic kidney disease. Lancet. 2007, 369( 9569): 1287-1301.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009, 20(9):1888-1893.
- Paul BM, et al. Evidence of a third ADPKD locus is not supported by re-analysis of designated PKD3 families. Kidney Int. 2014, 85(2): 383-392.
- Tess Harris. Is It Ethical to Test Apparently "Healthy" Children for Autosomal Dominant Polycystic Kidney Disease and Risk Medicalizing Thousands? Frontiers in Pediatrics. 2018, doi: 10.3389/fped.2017.00291.
Inquiry
