Bartter Syndrome
Bart syndrome (BS) was named after Bartter et al. first reported two cases of hypokalemia metabolic alkalosis, hyperaldosteronism, normal blood pressure, and renal histological examination showed hypertrophy of the paraglomerular organs in 1962. By the 1990s, more than 200 cases have been reported, and some patients may have a family history. The pattern is autosomal recessive inheritance. Clinical data shows that although the cases named after BS are characterized by the loss of kidney salt, hypokalemia metabolic alkalosis, increased blood renin and aldosterone levels, and normal blood pressure, they have other clinical manifestations and laboratory tests. Characteristics, so BS refers to a group of inherited renal tubular diseases characterized by hypokalemia and metabolic alkalosis. The nomenclature of BS alone can no longer satisfy the understanding of the pathogenesis and the guiding significance of clinical diagnosis and treatment. At present, BS is divided into the following types: (1) classic Bart syndrome (cBS); (2) Gitelman syndrome (GS); (3) neonatal Bart syndrome or hyperprostaglandin E syndrome (aBS/HPS).
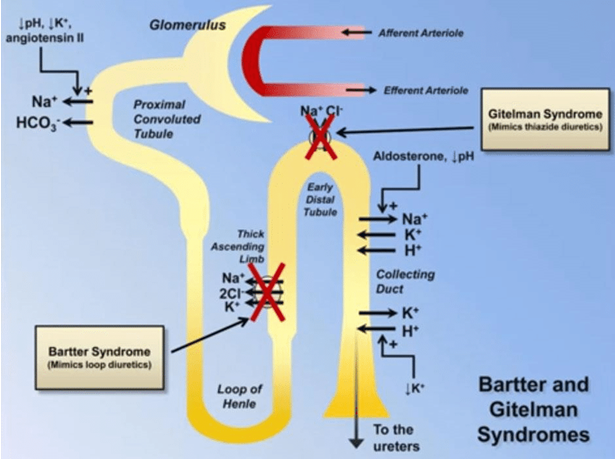
Figure 1.Illustration of Bartter syndrome, lose excess amounts of salt (sodium chloride) through their urine. (Zulkar Nain, 2016)
In recent years, the progress of molecular biology has begun to clarify the molecular genetics basis of different types of BS, providing greater help for clinical diagnosis and treatment. The glomerular filtration of various electrolytes is complete. The 20% filtered Na+ and K+ are reabsorbed in the ascending thick segment of the medullary loop. At the same time, with the absorption of Na+, 90% of the filtered Ca2+ is reabsorbed in a passive manner. The specific process and the role of the relevant ion channels have been elucidated: Each of the Na+, K+ and two Cl- in the thick section of the ascending branch of the renal tubular medullary loop enters the cell through the Na+-K+-2Cl- transport system on the top cell membrane of the luminal side of the renal tubular epithelial cell in the form of electrical neutralization . There is a Na+-K+ pump on the cell membrane at the bottom of the cell. The Na+ in the cell is transported out of the cell into the blood vessel. For every 3 Na+ pumped out, the K+ in the 2 interstitial blood vessels will enter the cell. This is an active process that requires ATP consumption, so such pumps are called Na+-K+ ATPase pumps. The K+ entering the cell mainly moves out of the cell via the K+ channel. This kind of channel molecule responsible for transporting K+ is called the extracellular myelin potassium channel (ROMK) because it was originally found to be expressed in the outer medullary part of the rat kidney tissue, and its molecular expression has been found in human kidney tissue, but it is still used. ROMK is distributed on the top and bottom of the renal tubular cells, so the K+ in the cells will enter the tubular lumen and the vascular lumen, respectively. The opening and closing of ROMK depends on the intracellular Ca2+ and ATP levels. When the Na+-K+ pump is active and the intracellular ATP is consumed, the K+ channel will be triggered to open, so some scholars call it the ATP-regulated K+ channel. The K+ recycling thus completed promotes Na+-K+-2Cl- to enter the cell through the top cell membrane, and maintains the positive charge on the lumen side, so that K+, Na+ can also enter the blood vessel through the intercellular space, and Ca2+ and Mg2+ also pass through the intercellular space is absorbed. The intracellular Cl- enters the interstitial blood vessels through the kidney-specific chloride channel (CLC-Kb) and K+/Cl- co-transport system located at the base of the cell membrane. There is also Na+/H+ exchange on the top cell membrane. The distal renal tubule and collecting duct reabsorb 12% filtered Na+. The distal renal tubules can be divided into a cortical part (called the cortical dilution section, which is the beginning of the distal tubule) and a medullary part (which is the posterior part of the distal tubule). Cortical renal tubular epithelial cells reabsorb NaCl through the Na+/Cl- common transport system of the luminal facial cell membranes. The medullary renal tubular cells have two types of cells: the main cells reabsorb Na+ and water and secrete K+; the inserted cells secrete H+ through H+/K+ exchange and reabsorb K+. Aldosterone can enhance the function of both cells. The dysfunction of different ion channels in the above sites is the pathophysiological basis of various types of BS, and causes their respective clinical characteristics.
Classical Barth Syndrome (cBS)
Initial pathological studies have found that renal tissue biopsy in children with cBS shows paraproximal hyperplasia. In recent years, Okada et al. also reported that their work showed that the number of renin-containing cells in the paraglomerular organs of BS patients increased significantly compared with normal controls. However, it is currently believed that this is not its primary lesion. The pathogenesis of cBS is related to the Cl-Kb channel defect causing Cl- reabsorption. It is precisely because of the Cl- transport disorder that affects the reabsorption of K+ that forms the clinical biochemical characteristics of cBS.
Variation of Bart Syndrome
The study found that the defect of the Na+/Cl- common transport system in the distal tubular cortex is the basis of variant pathogenesis of Bart syndrome. Na+ and Cl- reabsorption dysfunction leads to a slight lack of circulating blood volume, stimulating the renin-angiotensin-aldosterone system; causing the main cells of the distal renal tubule medulla to strengthen the reabsorption of Na+ and water, this process will expel K+; at the same time, the inserted cells secrete too much H+ and absorb K+. The secretion of H+ and absorption of Cl- become the cause of metabolic alkalosis.
Neonatal Bart Syndrome
It is now believed that the onset of neonatal Bart syndrome is related to defects in the membrane ion channel of the ascending thick segment of the myelin loop. Due to the absorption of Na+, K+, Cl- and Ca2+ by the renal tubules at this site, it further affects the osmotic pressure of the space around the renal tubules and reduces the reabsorption of water in the descending branch of the myeloid loop. As a result, urine containing large amounts of Na+, K+, Cl- and Ca2+ was discharged to the distal renal tubule. Na+ is reabsorbed in the form of Na+/ K+ exchange in the distal renal tubule. Obviously, the urine in the lumen is partially but not completely concentrated, but more K+ is wasted. The reason for the increase in prostaglandin E is currently believed to be secondary to the loss of water and electrolytes.
References
- Bartter F, et al. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemi c alkalosis. Am J Med. 1962, 33:811-828.
- Karolyi L, et al. The molecular genetic approach to “Bartter′s syndrome”. J Mol Med. 1998, 76:317-325.
- Amirlak I, DawsonKP. Bartter syndrome:an overview. Q JMed. 2000, 93:207-215.
- Zulkar Nain. 2016.GENETIC ANOMALIES IN KIDNEY. CreateSpace, an Amazon company.
Inquiry
