Benign Familial Neonatal Seizures
Benign familial neonatal convulsions (BFNC) is a rare idiopathic epilepsy syndrome, which is mainly manifested as febrile seizures during the neonatal period, and then disappears spontaneously within a few weeks or months. Its inheritance Dominant chromosome inheritance. The age of onset and disappearance of seizures is significantly different between patients within and outside the family. Although most BFNCs have a benign process, about 7% of patients still have language, cognitive and other neurological dysfunctions, and about 10% to 16% of patients will relapse to epilepsy in adolescence or adulthood. With individual development Imaging examination of the head of patients with obstacles can reveal abnormalities such as thinning of the corpus callosum, mild white matter atrophy and lateral ventricle enlargement. The study found that in patients with benign familial neonatal convulsions, there are pathogenic mutations in the potassium channel member genes KCNQ2 and KCNQ3.
In 1989, Leppert et al. conducted a linkage analysis of a Swiss BFNS family with 19 patients in four generations, and located the disease-causing gene on chromosome 20 q13.3, making BFNS the first chromosomal site to locate the relevant disease-causing gene. Since then, molecular genetics studies of other BFNS families have proven to be mostly at this site. In 1993, Lewis et al. performed a genome scan of a BFNS family of Mexican descent that included 14 patients in three generations, and located the disease-causing gene on chromosome 8 q24. In 1998, Singh et al. scanned the fetal brain cDNA library and identified the causative gene KCNQ2 co-segregated with a BFNS family phenotype at 20 q13.3. Then, in a BFNS family study, Charlier et al. found that it was linked to the No. 8 chromosome, and found a pathogenic mutation in the KCNQ3 gene. But at the same time, about 30% of the BFNS families conducted linkage analysis to prove that the disease-causing gene is not linked to the KCNQ2 or KCNQ3 site, and may be located at a new chromosomal site, suggesting that BFNS may have other disease-causing genes and have genetic heterogeneity.
The study found that KCNQ2 and KCNQ3 are widely overlapped and staggered in the brain. Immunohistochemical analysis showed that KCNQ2 and KCNQ3 can produce co-immunoprecipitation, indicating that KCNQ2/KCNQ3 has the function of co-localization and mutual coordination. They express together to form potassium channels of functional heteropolymerization, and involved in the formation of M channels. Further research found that the M channel is mainly composed of KCNQ2 and KCNQ3, followed by KC-NQ4 and KCNQ5, which are widely distributed in the nervous system, including brain regions related to seizures such as cerebral cortex, hippocampus and thalamus. The M channel current is a voltage-dependent slow activation/inactivation of outward potassium current, which plays an important role in adjusting the frequency of nerve impulse firing and maintaining and stabilizing neuronal excitability. This potassium ion current can be strongly suppressed by muscarinic, so it is called M current (IM).
KCNQ Family Protein Structure
The structure of the voltage-gated potassium channels encoded by the genes of the KCNQ family is quite similar. It is a tetramer composed of 4 identical or different α subunits, and there are 1 or more β subunits. The latter plays an auxiliary role on α subunit, and has the function of adjusting and stabilizing the channel. Each α subunit contains 6 hydrophobic α-spiral transmembrane domains (S1-S6), 1 K + selective P domain, and the amino terminal and carbon terminal located in the cytoplasm. S4 is a voltage receptor, regularly arranging positively charged amino acid residues. When hyperpolarized, S4 is located on the cytoplasmic side of the membrane; when the membrane is depolarized, the sensed electrical change will drive S4 to transfer from the inside to the outside of the membrane, resulting in activation and opening of potassium channels. The P domain contains the signal sequence GYG for the selective passage of potassium ions, and the P domains of the four α subunits together constitute the channel pore area. N-terminal studies have shown that it can affect the characteristics of the hole area. The C-terminal amino acid residues constitute the subunit interaction region (si), which determines the assembly of the KCNQ subunit. The si area contains two highly spiro motifs TCC1 and TCC2 composed of repeated hydrophobic amino acid residues.
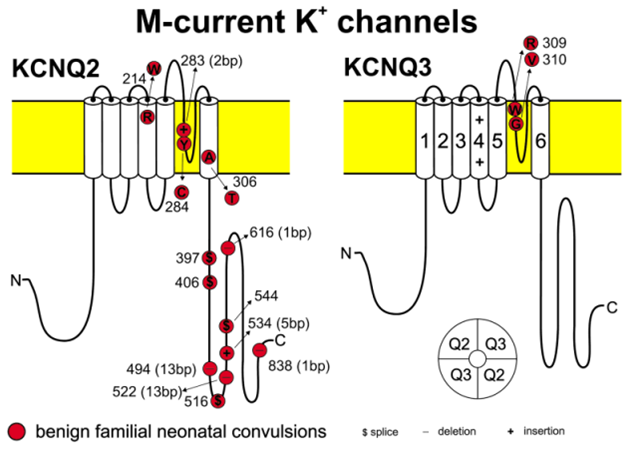
Figure 1. Proposed structure of the voltage-gated K channels KCNQ2 and KCNQ3 containing mutations causing benign familial neonatal convulsions. (HOLGER LERCHE, et al. 2001)
BFNC and KCNQ2, KCNQ3
It is known that the KCNQ2 mutation is mainly distributed in the S4 voltage receptor, C-terminal, membrane area and pore area. In the functional study of Xenopus oocytes, the mutant protein mainly affects the function of the M channel from the following aspects: S4 voltage receptor mutation reduces the voltage sensitivity of the M channel, slows its activation and accelerates inactivation, reduces the repolarization reserve of IM regulation, and reduces the ability to prevent neuronal discharge, resulting in increased susceptibility to epilepsy, but the maximum current And the expression level in the cell membrane has not changed; the C-terminal mutation can lead to functional KCNQ2 protein synthesis disorder, the effective expression level on the cell membrane is reduced, which in turn makes the M channel dysfunction, IM reduction, and weakening of the depolarization dependent potassium channel ; When the membrane area is mutated, although the KCNQ2 protein is normally expressed on the cell membrane, its function is weakened, and the IM formed with KCNQ3 can be reduced by 20% to 40% compared with normal; the pore area is where potassium ions pass, and the mutation in this area will This leads to a direct reduction of IM. KCNQ3 mutations are mainly distributed in the pore area, which reduces the maximum current of KCNQ2/KCNQ3 heteromeric channels by about 20% to 40%, but no effect on the membrane surface expression and voltage dependence of KCNQ3 protein has been found; in addition, BFNS family also found KCNQ3 mutation which is located in the junction region of the pore region and S6. It is speculated that this mutation may affect IM by changing the structure of the pore region or S6 transmembrane region, thereby affecting neuronal excitability.
As a result of the KCNQ2/3 gene mutation, as long as the maximum potassium ion flux is reduced by 20% to 30%, a haploinsufficiency of the ion channel is sufficient to cause BFNS. Mutant subunit don't affects the selectivity of the potassium channel, and the function of another normal subunit, so the occurrence of BFNS comes from the current reduction caused by the mutation of one of the two, rather than the complete IM disappearance or through the dominant-negative effect. Individual genotypes with KCNQ2/3 mutations are life-long, and the mechanism of spontaneous remission and age dependence of BFNS is currently unclear. It is speculated that it may be related to the age-dependent activation of KCNQ channel, that is, the KCNQ2/3 channel is expressed at a low level in the neonatal period, and the neonatal brain is not yet mature at this time, and the threshold for susceptibility to seizures is low. A slight decrease in IM caused by mutations can trigger convulsions, suggesting that the number of KCNQ2/3 channels during this period is directly related to proper control of subthreshold membrane potential; after the neonatal period, the number of KCNQ2/3 channels has been significantly increased due to increasing the number of open M channels, thereby avoiding excessive neuronal excitation, suggesting that the function of KCNQ2/3 channels after the neonatal period has an important role in the maintenance of neuronal membrane potential.
References
- Singh NA,et al.A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998,18 (1): 2 5-2 9.
- Kurata HT, et al.Amino-terminal determinants of U-type inactivation of voltage -gated K + chan- nels. J Biol Chem. 2002, 277(32): 29045-29053.
- Schwake M, et al.Structural determinants of M -type KCNQ ( Kv7 ) K + channel assembly. J Neurosci. 2006, 26(14): 3757-3766.
- Singh NA, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spec- trum. Brain, 2003, 126(Pt12): 2726-2737.
- Wang HS, et al. KCNQ2 and KCNQ3 po-tassium channel subunits: molecular correlates of the M -channel. Science, 1998, 282(5395): 1890-1893.
- HOLGER LERCHE, et al. Ion Channels and Epilepsy. American Journal of Medical Genetics. 2001, 106:146-159.
Inquiry
