Brugada Syndrome
The Brugada Syndrome (BrS) is a rare autosomal dominant genetic disease. The disease has a normal heart structure, but once the onset will have serious consequences, the main clinical manifestations are cardiogenic syncope, sudden death without organic heart disease, and the electrocardiogram is characterized by the V1~V3ST segment in a cape shape or saddle shape elevated. BrS has a tendency to cause sudden death due to rapid polymorphic ventricular tachycardia and ventricular fibrillation.
Brs' Basics of Genetics
So far, 19 gene mutations have been identified as related to the Brugada phenotype. Among them, the earliest and most studied gene is SCN5A. So far, hundreds of SCNSA mutant genes have been found to be related to Brugada syndrome, most of which are deletion mutations. These mutations lead to a decrease in sodium influx or calcium influx or an increase in potassium outflow, causing the current to move outward in the early stages of AP.
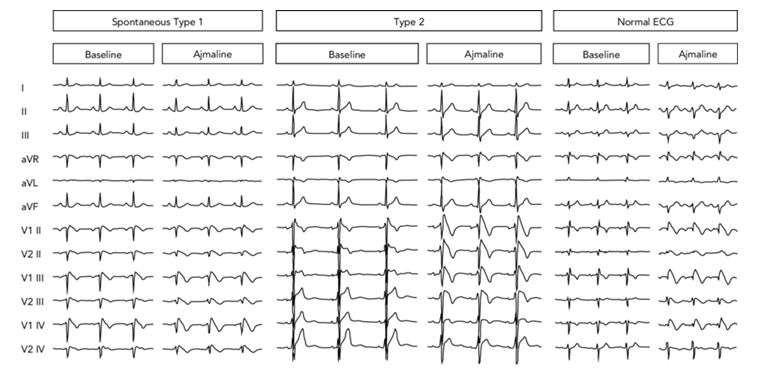
Figure 1. Electrocardiographic Pattern of Brugada Syndrome.(Carlo Pappone. et al., 2019)
Mutations That Cause Loss of Sodium Channel Current Function
SCN5A
The first gene associated with BrS is SCN5A, which is the gene encoding the alpha subunit of the voltage-gated cardiac sodium channel (Nav1.5). To date, more than 300 BrS-related mutations in SCN5A have been described, accounting for the majority of cases with positive BrS genotypes, but only accounting for 11% to 28% of previously identified BrS patients. Some of these mutations have been functionally expressed and appear to cause loss of INa function.
GPS1-L
In addition to SCN5A, Weiss et al. described a new BrS-related locus close to SCN5A. This locus was later identified as a GPD1-L gene encoding a 3-phosphoglycerol dehydrogenase-like protein. It has been found that it has a close structural and functional correlation with NaV1.5. Through GP1-L-dependent Nav1.5 phosphorylation, the impaired enzyme activity ultimately leads to a reduction in the activity of INa.
SCN1B
SCN1B gene encodes the auxiliary NaVβ1 subunit of the voltage-gated cardiac sodium channel. It was first determined by Watanabe et al. to cause loss of peak INa function. In subsequent studies, co-expression of mutant SCN1B with WT-SCN5A and WT-KCND3 induced a 55.6% reduction in the peak value of INa and a 70.6% increase in the function of Ito, indicating that in these BrS cases, the transient outward potassium current Elevation is its main pathogenesis.
SCN3B
Hu et al. reported a missense mutation in SCN3B, which encodes the Navβ3 subunit of myocardial sodium channels, which leads to a decrease in peak INa current density, accelerated inactivation, and slowed down reactivation. Studies have shown that mutations in this subunit impair intracellular transport and cellular expression of the sodium channel in the heart.
SCN2B
The SCN2B gene encodes the β2 subunit of the atrial sodium channel. Due to the mutation of this gene, the surface expression of Nav1.5 cells is reduced, resulting in a significant decrease in sodium current density. In addition, BrS mutations caused by the loss of sodium channel current function also include SCN10A, HEY2, FGF12, PKP2, RANGF, and SLMAP.
Mutations That Cause Loss of Calcium Channel Current Function
CAC-NA1C
The mutation in CACNA1C caused a loss of function, which encodes the alpha subunit of the human L-type voltage-gated calcium channel, and research results have shown that the mutation is related to many cases of BrS.
CACNB2B
The gene encoding the beta subunit of CaV1.2 is involved in the regulation and intracellular transport of ICal. Because of the sudden change of function caused the loss of ICal function, it was proved to be related to BrS.
CACNA2D1
CACNA2D1 encoding voltage depends on the α2δ subunit of the calcium channel, and has similar functional characteristics as Cavβ2.144. According to reports, the mutation of this gene may cause short QT syndrome (SQTS), idiopathic ventricular fibrillation, early repolarization syndrome (ERS) and BrS.
Mutations that lead to enhanced potassium channel current function
KCNEE3
The mutation in KCN3 (MiRP2) has been shown to be related to BrS. The function of MiRP2 is to regulate several cardiac potassium currents, including Ito and IKs. The joint expression of KCNE3 mutation and WT-KCND3 leads to an increase in the function of Ito.
KCND3
The function of KCND3 mutation is also related to BrS and it is shown that it can be increased by ITO.
SCN1B
The BrS-related mutation in the SCN1B gene, which encodes the auxiliary NaVβ1 subunit of the voltage-gated cardiac sodium channel, in addition to the reduction of INa, it can also increase Ito when co-expressed with WT-KCND3.
KC-NJ8
It has been reported that the mutation of KCNJ8 encoding Kir6.1 will lead to an increase in the function of IK-ATP. Mutation leads to the enhancement of the notch of the action potential and the depression of the platform. The shortening of the duration of the AP leads to the BrS phenotype or SQTS phenotype.
ABCCC9
Sudden mutation in ABCCC9, encoding SUR2A, adenosine triphosphate (ATP) binding transport of IK-ATP channel
It has recently been identified as a causative agent of BrS. The mutation of ABCCC9 can reduce the sensitivity of ATP receptor complexes, because the inhibitory effect of ATP is reduced, which leads to an increase in function.
References
- Carlo Pappone, Vincenzo Santinelli. Brugada Syndrome: Progress in Diagnosis and Management. Arrhythmia & Electrophysiology Review. 2019, 8(1):13-8.
- Kapplinger JD, et al.; An inter-national compendium of mutations in the SCN5A encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm,2010,7 :33- 46.
- Hu D, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014,64:66-79.
- Antzelevitch C, Patocskai B. Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects. Curr Probl Cardiol. 2016;41(1):7-57.
- Sieira J, et al.; Pathogenesis and management of Brugada syndrome. Nat Rev Cardiol. 2016;13(12):744-756.
Inquiry
