Catecholamine-Sensitive Pleomorphic Ventricular Tachycardia
Catecholamine-sensitive pleomorphic ventricular tachycardia (CPVT) is a primary cardiac electrical disorder that occurs mostly in adolescents with no organic heart disease and a normal QT interval. Characterized by bidirectional or polymorphic ventricular tachycardia (VT) during exercise or emotional agitation, leading to syncope and sudden death. The disease was first reported by Reid et al. in 1975. Since then, there have been multiple cases reported, but the diagnosis name is different. In 1995, Leenhardt et al. reported the first larger series of case studies, describing in detail the clinical manifestations of the disease, and officially named the disease with this clinical feature CPVT. Subsequently, more and more people realized that CPVT is an important cause of sudden death in patients with non-organic heart disease, and they have also updated their etiology, pathogenesis, and diagnosis and treatment strategies.
CPVT has obvious family aggregation, there are two forms of autosomal dominant inheritance and recessive inheritance, which are related to mutations of genes RyR2 and CASQ2, respectively. About 50% of patients have RyR2 gene mutations, and about 1% to 2% have CASQ2 gene mutations. In addition, there are reports that mutations in the anchoring protein B gene can also cause CPVT, but other studies have yet to confirm it. The RyR2 channels expressed by the RyR2 gene are distributed on the sarcoplasmic reticulum of cardiomyocytes, and play an important role in the balanced regulation of cytosolic free Ca2+ concentration. The CASQ2 protein expressed by the CASQ2 gene is involved in the buffering and storage of Ca2+ in the sarcoplasmic reticulum. The complex formed by CASQ2 and RyR2 is necessary for sarcoplasmic reticulum to release Ca2+. The researchers found that in sympathetic excitement, sympathetic excitement can abnormally increase the opening of the mutant RyR2 channel, leading to diastolic Ca2+ leakage, intracellular Ca2+ overload, and induced depolarization after delay. A study of the CASQ2 gene mutation found that the mutated CASQ2 protein can reduce the ability of the myocardial cell sarcoplasmic reticulum to store and release calcium ions. When it is subjected to pacing electrical stimulation or exposure to norepinephrine solution, membrane potential appears Vigorously oscillated with delay after depolarization. Recently, the study of computer simulation models also confirmed that abnormal function of RyR2 channel and CASQ2 protein can affect the calcium homeostasis in cardiomyocytes, which in turn can cause delayed depolarization of the cells.
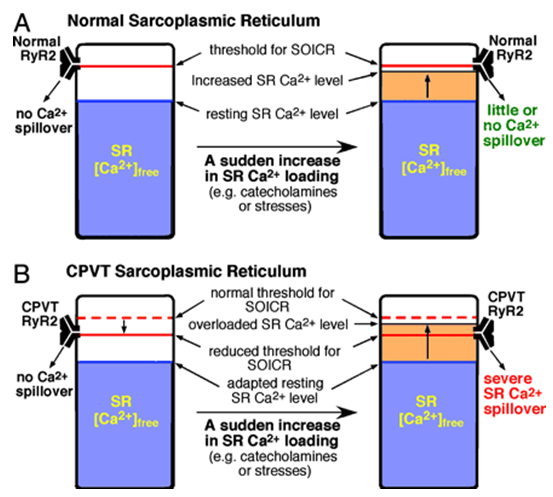
Figure 1. A proposed mechanism for CPVT associated with RyR2 mutations.(Dawei Jiang, et al., 2004)
At the tissue level, Nam et al. in the isolated canine ventricular tissue caused calcium leakage in the sarcoplasmic reticulum through perfusion of caffeine to simulate the pathophysiological state of CPVT. In the experiment, they found that the delay induced by isoproterenol mostly originated in the epicardium, which in turn caused an increase in the transmural dispersion of repolarization, and eventually led to CPVT-like VT or ventricular fibrillation (VF). For in vivo studies, Cerrone et al. transferred the mutated RyR2 gene into the heart of mice, and recorded the bidirectional VT characteristic of CPVT under the stimulation of epinephrine. Knollman et al. found in CASQ2 protein-deficient mice that both isoproterenol and exercise test can induce CPVT-like bidirectional and polymorphic VT. Paavo la et al. used a one-way action potential mapping catheter to map 15 CPVT patients with RyR2 gene mutations and found that under intravenous drip adrenaline, 4 cases had delayed depolarization and accompanied by Ventricular premature beats, and no similar phenomenon was found in the control group.
Most scholars currently believe that abnormal RyR2 channel or CASQ2 protein-induced depolarization after delayed sympathetic excitement may be the mechanism of CPVT. However, there are multiple mutation sites in CPVT patients, and whether they all show the same pathogenesis remains to be proved by research. In addition, some patients do not have gene mutations, and little is known about their causes and pathogenesis. Due to the lack of in vivo studies, it is unclear how the delayed depolarization in the whole heart and how local triggering activities evolve into bidirectional or polymorphic VT.
References
- Reid DS, et al. Fitzgerald, Bidirectional tachycardia in child study using his bundle electrography. Br Heart J. 1975, 37:339.
- Leenhardt A, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91(5):1512-1519.
- Lehnart SE, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004, 109:3 208.
- Dawei Jiang, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+release (SOICR).Proceedings of the National Academy of Sciences. 2004, 101(35): 13062-13067.
Inquiry
