Congenital Hyperinsulinemia
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infants, so CHI is also known as persistent hyperinsulinemia hypoglycemia in infants. In 1954, MacQuarriet first introduced infantile idiopathic hypoglycemia Report the disease. CHI is a severe hypoglycemia caused by the continuous inappropriate secretion of insulin by β-cells of the islets. Its clinical features are high insulin-induced hypoglycemia in infancy, often with low ketone body and hypofatty acidemia, and its hypoglycemia is often difficult to correct, leading to neurological complications (especially hypoglycemic encephalopathy), which can cause disability and even lethal. The etiology and pathogenesis of this disease are not yet fully understood, and the estimated incidence in the total population is 1 /30000 to 1/50000. In the close marriage group, the incidence is as high as 1/2500. At present, people recognize that this is a group of inherited ion channel diseases. Several related genes have been identified: sulfonylurea receptor (SUR1) gene, The inward rectifying potassium channel (Kir) gene, glutamate dehydrogenase (GDH) gene, glucokinase (GK) gene, short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) gene, etc.
So far, among the reported genes that cause CHI, the inactivating mutations of KCNJ11 and ABCC8 genes are the most common causes of CHI, accounting for about 40% to 45% of CHI; other gene mutations account for about 5% to 10% of CHI patients ; The remaining 50% of CHI patients have unknown disease genes.
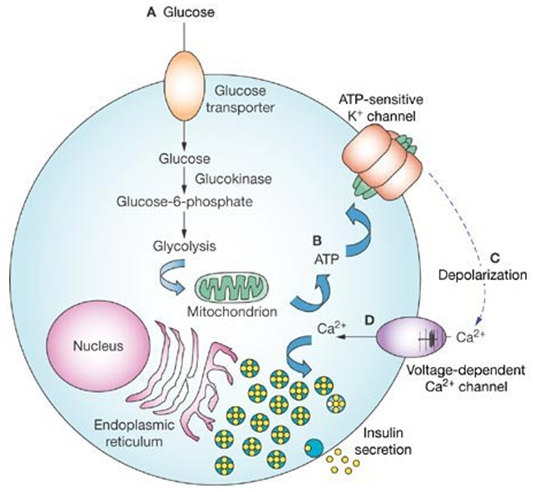
Figure 1.Glucose-stimulated insulin secretion in pancreatic β-cells. (De León DD, et al.; 2007)
KATP CHANNEL
The ATP-sensitive potassium channel (KATP channel) on islet β cells is composed of two subunits, SUR1 and Kir6.2. SUR1 is encoded by the ABCC8 gene, and Kir6.2 is encoded by the KC-NJ11 gene, both of which are located on chromosome 11p15.1. Among them, SUR1 is the regulatory subunit of the KATP channel, and Mg-ADP, diazoxide, sulfonylurea, and gleneide drugs can be combined with it, thereby affecting the KATP channel. SUR1 can sense changes in the concentration of ATP/ADP in the cell, regulate KATP channels, allow potassium ions to enter the cell from a high concentration, and maintain the polarization state of the cell membrane. In a physiological state, glucose enters islet β cells through glucose transporter 2 (GLUT2), and is catalyzed by glucokinase to produce glucose 6-phosphate (G-6-P). G-6-P continue to be hydrolyzed or enter the tricarboxylic acid cycle to produce a large amount of ATP. And the increase of [ATP]/[ADP] will affect SUR1, making Kir6.2 Closed, causing depolarization of the cell membrane, causing voltage-gated calcium ion channels to open, forming calcium influx, and finally causing the insulin β cells to release insulin. SUR1 and Kir6.2 Inactivation will lead to the continuous closure of KATP channels and continuous depolarization of β-cell membranes, which ultimately leads to dysregulation of insulin secretion. The recessive inactivating mutations of the ABCC8 and KC-NJ11 genes usually cause more severe CHI.
Glutamate Dehydrogenase
Glu-dehydrogenase-related CHI, also known as hyperinsulin/hyperammonemia syndrome, is the second most common type of CHI. It shows autosomal dominant inheritance or sporadic. Under normal circumstances, the protein in food is broken down into amino acids and absorbed by the intestine. As an alternative path for glucose energy supply, some amino acids, such as glutamic acid, can be converted into the tricarboxylic acid cycle after conversion to produce ATP, which increases [ATP]/[ADP], and then the KATP channel is closed, causing insulin to be secreted by pancreatic β cells. The key enzyme for glutamic acid to enter the tricarboxylic acid cycle is glutamate dehydrogenase (GDH), which catalyzes the hydrolysis of glutamic acid to produce α-ketoglutarate and ammonia, and α-ketoglutarate can directly enter tricarboxylate Acid cycle. GDH is mainly distributed in pancreas, liver, kidney, brain and other organs. In addition to catalyzing the deamination of glutamic acid, it can also catalyze the deamination reaction of other amino acids (leucine, valine, etc.). Under normal circumstances, GDH can be activated by ADP and leucine, and inhibited by GTP and ATP.
The GLUD1 gene encoding GDH is located on chromosome 10p23. The mutation usually occurs in the GTP allosteric binding region of GDH. GLUD1 gene mutation makes GDH less sensitive to the allosteric inhibitor GTP, resulting in increased GDH activity, which increases the amount of α-ketoglutarate produced by glutamic acid, increases [ATP]/[ADP], and excessive release of insulin. Another characteristic of GDH-CHI is elevated blood ammonia. GDH with enhanced activity consumes glutamic acid and generates a large amount of ammonia; on the other hand, the reduction of glutamic acid leads to a reduction of N-acetylglutamic acid, which in turn affects carbamoyl phosphate synthetase, impairs urea synthesis, and eliminates ammonia Weaken. The blood ammonia concentration is usually 3 to 5 times the normal value, but the neurological symptoms of hyperammonemia will not be shown clinically. GDH-CHI patients have normal birth weight and mild hypoglycemia, mainly caused by long-term fasting or protein diet. Because leucine is an activator of GHD, patients often get sick when they switch from low-protein breast milk to infant formula. GDH-CHI diazoxide is effective and can prevent hypoglycemia by restricting protein intake (especially leucine-rich protein).
Glucokinase
Glucokinase (GCK) is encoded by the GCK gene located on chromosomes 7p15.3 ~ p15.1. GCK is the rate-limiting enzyme for glucose metabolism in pancreatic islet β cells. When the GCK gene is activated and mutated, the affinity of GCK for glucose increases, the rate of G-6-P production increases, and [ATP]/[ADP] increases, resulting in inappropriate insulin secretion. A total of 15 kinds of GCK gene activation mutations are currently found, which are dominant inheritance or sporadic. Among them, V455M is a more common type of mutation. The reported mutations are all caused by increased affinity for glucose, resulting in increased enzyme activity. There is no evidence that GCK gene overexpression is the cause of CHI, and its mutation sites are mainly concentrated in the allosteric activation region. Because the activation of GCK mutations lowers the threshold of insulin secretion, most of GCK-CHI shows mild fasting and postprandial blood sugar reduction, and blood insulin levels rise slightly to moderately. In general, the clinical manifestations and development process of GCK-CHI patients are quite different, and even the clinical manifestations of CHI caused by the same mutation in the same family are different, suggesting that the mechanism of GCK regulation is very complicated. The age of onset of this disease spans a wide range, from infancy to adulthood, and most GCK-CHI diazoxide treatments are effective.
Hydroxyacyl-CoA Dehydrogenase
Fatty acid β oxidation is the main way to supply energy to adipose tissue. The process is to produce acetyl-CoA through a series of oxidation and cleavage of linear fatty acids. Acetyl-CoA and oxaloacetate enter the tricarboxylic acid cycle, producing ATP. The hydroxyacyl-CoA dehydrogenase (HADH) encoded by the HADH gene is a key enzyme for fatty acid β oxidation. The HADH gene is regulated by transcription factors such as Foxa2 and can be expressed in a variety of tissues, among which the Langerhans cells in the islets are most expressed. The mechanism by which HADH gene inactivation mutation causes CHI is unclear. Studies have shown that HADH may be an inhibitor of GDH. HADH gene inactivation mutation leads to enhanced GDH activity, causing hyperinsulinemia. This can also explain the leucine-induced hypoglycemia in patients with HADH-CHI; but unlike GDH-CHI, HADH-CHI does not have hyperammonemia. Currently reported patients are all recessively inherited, and some of them have urine 3-hydroxyglutarate and blood 3-hydroxybutyryl elevation of carnitine. HADH-CHI has different clinical manifestations, which can be manifested as severe hypoglycemia in newborns or mild hypoglycemia in later stages.
In addition, it also includes hyperinsulinic hypoglycemia caused by anaerobic exercise or pyruvate load caused by mutations in the SLC16A1 gene; mutations in the gene encoding mitochondrial uncoupling protein 2 (UCP2) cause the leakage of protons, causing the coupling of oxidative phosphorylation in the mitochondria, ATP synthesis is reduced, and energy is lost in the form of heat. Mutation of UCP2 gene inactivation leads to a decrease in UCP2 uncoupling effect and an increase in ATP synthesis, which increases [ATP]/[ADP] and excessive insulin release. There are still unexplained genetic mutations that cause CHI, including the HNF4A gene that encodes hepatocyte nuclear factor 4α, and the HNF1A gene that encodes the hepatocyte nuclear factor 1α.
References
- Kapoor R, et al.; Clinical and molecular charac- terisation of 300 patients with congenital hyperinsulinism. Europe- an Journal of Endocrinology. 2013, 168(4): 557- 564.
- Hussain, K. Congenital hyperinsulinism. Seminars in Fetal and Neonatal Medicine. 2005, 10 (4): 369-376.
- Rahman, Sofia A.; et al.; Molecular mechanisms of congenital hyperinsulinism. Journal of Molecular Endocrinology. 2015, 54 (2): R119-R129.
- De León DD, et al.; Mechanisms of Disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007, 3(1):57-68.
Inquiry
