Congenital Insensitivity To Pain
Congenital insensitivity to pain is a genetic disorder of sensory autonomic nervous system. In patients with this type of disease, the transmission of pain is blocked, that is, pain sensation is dull or lost, and may be accompanied by anhidrosis, but the intelligence and sensory abilities such as cold and heat, vibration, and movement perception develop normally. Complete loss of pain means loss of alertness to harmful stimuli. A life system with complex regulatory functions will not easily lead to self-destructive "painless". It is estimated that including those who have not been diagnosed, there may be fewer than 40 painless and sweat-free patients in the world. Pain-free and sweat-free disease is still an unsolved mystery in medical circles all over the world.
The clinical manifestations of the patients are almost the same. When infants and young children start to crawl or walk, they often find that they do not respond to normal painful stimuli, even acupuncture. Frequent falls and burns due to analgesia result in fractures and dislocations, avascular necrosis, ulcers, suppurative infections, osteitis, scar formation, obvious colloids and charcot joints. In additon, facial injuries, lip and tongue Ulcers, corneal opacity. These children like to invade other children, and suffer many painless traumas. The intelligence is generally average, and some are mentally retarded. Unexplained fever, no or less sweat.
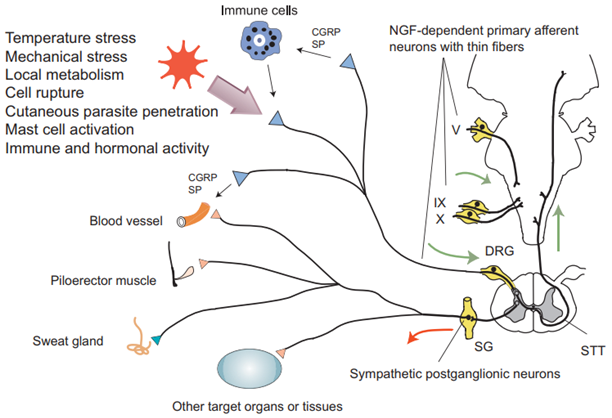
Figure 1. Patients with congenital insensitivity to pain with anhidrosis lack NGF-dependent primary afferent neurons with thin fibers and autonomic sympathetic postganglionic neurons.(Yasuhiro Indo, et al.; 2018)
Pathogenesis of congenital analgesia
All causes of CIP affect nociceptors (specific peripheral sensory neurons) and cause nociceptors or nociceptor neurodevelopmental failure. Congenital insensitivity to pain is an extremely rare phenotype, and the exact proportion of individuals with pathogenic variants in each gene in the entire population is unclear.
CIPA was first described in 1932. In 1983, Dyck et al. named various diseases associated with analgesia as hereditary sensory and autonomic neuropathy. Indo et al. first discovered and identified NTRK1 located on lq21~q22 as its pathogenic gene in 1996. NTRK1 gene is associated with familial medullary thyroid carcinoma, hereditary sensory and autonomic neuropathy type 4 (also known as congenital pain insensitivity with anhidrosis). Among them, the hereditary mode of familial medullary thyroid cancer is autosomal dominant inheritance, and hereditary sensory and autonomic neuropathy type 4 are inherited autosomal recessive. NTRK1 gene is a part of human trk oncogene derived from colon cancer. It contains 17 exons and encodes 790 or 796 amino acids. The gene encoded tyrosine kinase receptor A (TRKA) is the high-affinity receptor of nerve growth factor (NGF) , NGF-TRKA system regulates the growth and development of peripheral and central neurons, maintains the survival of neurons, and plays a key role in pain, itching and inflammatory infections. Because NTRK1 gene mutation affects the encoding TRKA, the signal transduction pathway of NGF is blocked, axon apoptosis, and sensory neuron developmental defects, resulting in a complete lack of unmyelinated nerve fibers and small cells in the dorsal root ganglia and skin. The myelinated nerve fibers in the skin lose the control of the autonomic sympathetic neurons in the sweat glands, causing loss of superficial pain and no sweating, and then leading to hereditary sensory and autonomic neuropathy.
Mutations in the SCN9A gene cause congenital insensitivity to pain. The SCN9A gene participates in the formation of a sodium channel part (α subunit), which is called NaV1.7. Sodium channels transport positively charged sodium atoms (sodium ions) into the cell and play a key role in the cell's ability to generate and transmit electrical signals. NaV1.7 sodium channels are found in nerve cells called nociceptors, which can transmit pain signals to the spinal cord and brain. The NaV1.7 channel also exists in the nerve cells in the nasal cavity among the olfactory sensory neurons, and transmits olfactory-related signals to the brain. Mutations in the SCN9A gene that cause congenital insensitivity to pain lead to the production of non-functional alpha subunits that cannot bind to the NaV1.7 channel. As a result, the formation of NaV1.7 channels is limited. The lack of NaV1.7 channel will weaken the transmission of pain signals from the injured site to the brain, thereby making the affected person less sensitive to pain. The loss of this channel in olfactory sensory neurons may impair the transmission of smell-related signals to the brain, leading to insomnia.
In addition, genes that trigger congenital insensitivity to pain may also involve the functions of genes such as CLTCL1, NGF, PRDM12, SCN11A, and ZFHX2. Therefore, the cause of congenital analgesia is very complicated, and its specific pathogenesis needs to be further explored.
References
- Chen Y, et al.; Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2019;44(10):1203-1208. doi:10.11817/j.issn.1672-7347.2019.190390.
- Schon KR, et al.; Congenital Insensitivity to Pain Overview. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993.
- Yasuhiro Indo, et al.; NGF-dependent neurons and neurobiology of emotions and feelings: Lessons from congenital insensitivity to pain with anhidrosis. Neuroscience and Biobehavioral Reviews. 2018, 87: 1-16.
Inquiry
