Congenital Stationary Night Blindness
Congenital static night blindness(CSNB) is a type of hereditary ophthalmological disease that mainly damages the function of the retinal rod system. Its clinical characteristics are non-progressive night blindness after birth, with normal vision and field of vision, no fundus changes, and the condition remains unchanged for life. Its inheritance mode is in line with Mendelian inheritance. At present, autosomal dominant inheritance, autosomal recessive inheritance and X-linked recessive inheritance have been reported. Among them, autosomal dominant inheritance is more. Due to the obvious genetic heterogeneity and allelic heterogeneity, its etiology and pathogenesis are very complicated, and it is still unclear, and there is no specific treatment to cure it. Therefore, elucidating the pathogenesis of such diseases at the molecular level has become one of the frontier research topics in the international ophthalmology community in recent years.
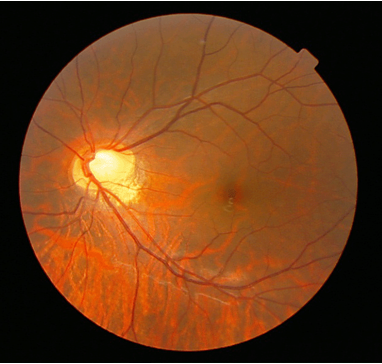
Figure 1. fundus of a patient with congenital stationary night blindness.(Inna Zolnikova, et al.; 2006)
Although autosomal dominant congenital static night blindness (ADCSNB) was discovered earlier and has a large number of literature reports, its research is very slow. The initial research mainly focused on clinical diagnosis. Because only the analysis of medical history, symptoms and signs could not identify the molecular defects and gene products of genes, the pathogenesis is still unclear. In the past 10 years, with the rapid development of genetic engineering and molecular biology technology and its wide application in the field of ophthalmology, the research on congenital static night blindness has also made great progress. Up to now, 3 genes related to ADCSNB have been found, they are rhodopsin gene (RHO), phosphodiesterase β subunit gene (PDE6B), guanine nucleotide binding protein α transduction active peptide 1. These genes have been cloned, and the properties of related protein mutants have been further studied through in vitro experiments, and new understanding of the pathogenesis of ADCSNB has also been obtained.
RHO Gene
The RHO gene is located on human chromosome 3q21-q24, with 4 introns and 5 exons, and the gene is 6706 bp in length. The encoded product is rhodopsin composed of 348 amino acids. The protein is composed of 11-cis-retinal and opsin, and its structure is very conservative. It contains a seven-span membrane core domain, 3 intracellular domains and 3 extracellular domains. Rhodopsin is exclusively expressed in rod cells. It is a highly specific G protein-coupling receptor that crosses the double phospholipid layer of cells and can transmit various extracellular signals. It belongs to the photoreceptor visual phototransduction system The receptors in the media can stimulate light cascades, amplify stimulus signals and cause hyperpolarization of photoreceptor cells and the release of neurotransmitters at synapses.
PDE6B Gene
The PDE6B gene is located on chromosome 4p16.3. The full length of the gene is 45170 bp with 21 introns and 22 exons. The encoded product is a phosphodiesterase β subunit consisting of 854 amino acids. This protein belongs to the membrane effector in the photoreceptor visual photoelectric transduction system, and regulates the concentration of the second messenger cyclic guanylic acid (cGMP) in the cell.
In 1994, Gal et al. studied the Danish Rambusch family and found that the first base of codon 258 of PDE6B gene‘s exon 4 in ADCSNB patients had a point mutation form C to A, which caused the histidine to be mutated to aspartyl. Gal et al. believe that this mutation activates cyclic guanylate phosphodiesterase in rod cells under dark adaptation, which leads to night blindness.
GNATI Gene
The GNAT1 gene, located at 3p21, has a total length of 4907bp, with 8 introns and 9 exons. Alternate shearing of GNATI gene produces two transcription products. The GNAT1 encoded by it has 350 amino acids, which belongs to the Gt protein in the G protein superfamily. It is the converter in the photoreceptor visual photoelectric transduction system and is coupled with rhodopsin, and then they activate the phosphodiesterase as an effector, which strengthens the hydrolysis of cGMP in the outer membrane of the rod cell. The hydrolysis of cGMP induces the decreasing of cGMP. Therefore, the decrease of cGMP combined with Na+ channels on the outer membrane of rod cells leads to the closure of Na+ channels and the decrease of Na permeability, which causes hyperpolarization of the photoreceptor cell membrane, and transmits light-induced visual signals to secondary retinal neurons. Therefore, if the GNATI gene is mutated, the light-induced visual signal cannot be transmitted and blindness.
References
- Barnes CS, et al.; A distinctive form of congenital sationary night blindness with cone on-pathway dysfunction. Ophthalmology. 2002,109: 575-583.
- Menon ST, et al.; Rhodapsin: struclural basis of Menon ST, Han M, SakmarTP. Rhodapsin: struclural basis of nalecular physiology. Physiol Rev. 2001, 81(4): 1659-1688.
- Gal A, et al.; Heterozygous mutation in the rod cGMP phosphodiesterase β-subunit gene in autosomal dominant stationary night blindness. Nat Genet.1994, 7: 64-68.
- Calvert PD, et al.; Phototransduction in transgenic mice after targeted deletion of the rod transducin α-subunit. Proc. Natl Aead Sci USA. 2000, 97(25):13913-13918.
- Inna Zolnikova, et al.; The clinico-functional and biomechanical aspects of pathogenesis, diagnostics and treatment of congenital myopia: review of the literature and the analyses of the native data. 2016, DOI: 10.18821/1993-1859-2016-11-3-149-157
Inquiry
