Cystic Fibrosis
Cystic fibrosis (CF) is the most common lethal autosomal recessive disease. It is caused by a gene mutation on autosome No. 7 that encodes the cystic fibrosis transmembrane transcription regulator (CFTR) protein. Inherited CF disease causes chronic lung infection and deterioration of lung function, leading to death. The common manifestation of CF is that there is a salty taste in sweat, the metabolism is not vigorous, and there are a variety of fecal problems. CF is mainly considered to be a chronic disease, and respiratory infection is the main cause of its incidence. In 5% to 13% of infants with CF, symptoms such as insufficient nutrition, insufficient blood protein, edema, and anemia may also occur.
The Structure and Function of CFTR
CFTR is a cyclic adenosine monophosphate-dependent chloride channel protein, a member of the adenosine triphosphate (ATP) binding protein family, and contains two transmembrane domains and two nucleotide binding domains (NBDs). CFTR contains a special regulatory domain and is the only channel protein in this family. The two NBDs in CFTR form a head-to-tail dimer, forming two ATP binding sites on the contact surface, one of which is formed by the Walker A motif of NBD1 and the LSGGQ motif of NBD2 (site 1), the other is formed by the Walker A motif of NBD2 and the LSGGQ motif of NBD1 (site 2). In addition, there is an AMP binding site on NBD2. The surface of the respiratory tract is attached with a mucus-ciliary scavenging system, which is mainly composed of columnar epithelial cells and cilia, mucus layer, serous layer, goblet cells and serous cells. The mucous gland is the main site of CFTR expression. In the respiratory epithelium, CFTR drives the secretion of fluids and electrolytes, and secretes mucin, sugar complexes, antibacterial agents, etc. from the mucous glands and flows into the respiratory tract, so it plays an important role in mucociliary clearance and self-defense.
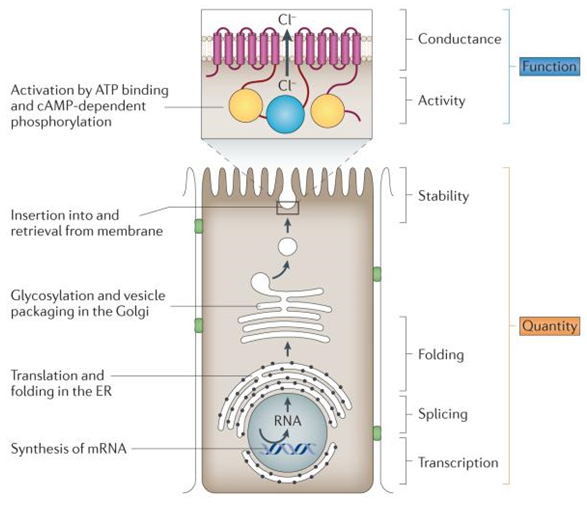
Figure 1. Molecular consequences of variants in CFTR(Cutting GR, 2015)
Pathogenesis of CF Lung Disease
CFTR mutation is the main cause of CF. Clinical studies have found that CF tracheal epithelial cells have two defects in the process of transepithelial ion transport: the permeability of the epithelial membrane chloride ions is greatly reduced, and they do not respond to cyclic adenosine phosphate agonists. The permeability of epithelial sodium ions is greatly improved. In CF, the functional damage of CFTR leads to the obstruction of the mucous glands, which prevents their secretions from flowing into the tracheal lumen. The mucus cilia in the trachea cannot be cleared, leading to chronic bacterial infections and tracheal inflammation, and ultimately to lung function damage. Two different hypotheses have been established to explain the pathogenesis of CF lung disease: the high salt hypothesis and the low volume hypothesis. The hypothesis of high salt is mainly based on the regulation of C l-channel by CFTR. The mucus layer and serous layer in the normal mucus-ciliary scavenging system are called the respiratory surface fluid layer (ASL). These two layers of liquid can effectively regulate cilia movement and mucus removal. The mutation of CFTR leads to the failure of the transport of saline that it drives. Although AS absorbs a lot of water, the concentration of salt continues to increase, causing the antibacterial substances in it to inactivate, thereby damaging the self-defense system in the respiratory tract.
The hypothesis of low volume is mainly based on the effect of CFTR on the epithelial sodium channel (ENaC). The functional damage of CFTR cannot inhibit the absorption of liquid and electrolyte by ENaC. This accelerated absorption greatly reduces the volume of ASL. The dehydration of the mucus layer causes the fluid cilia to not be removed well. In normal lungs, the height of the serous layer is regulated by hormones, combined with the absorption of Na+ by ENaC in the epithelial cell membrane, the Cl- secretion activated by CFTR and the Cl- channel activated by Ca2+, which keep the height of the serous layer is maintained at 7 μm; when the CFTR function is lost, the absorption of Na+ is no longer restricted, and the secretion of Cl- fails, both of which cause the volume of ASL to decrease. If there is no compensation mechanism, the mucus clearance phenomenon will disappear within 48 hours. However, the decrease in ASL height will lead to an increase in the release of epithelial ATP, which activates the intracellular purinergic receptor P2Y2 and inhibits the absorption of Na+, and activated the secretion of Cl- regulated by Ca2+. In a steady state, this mechanism can partially compensate for the volume of ASL; in the presence of external pressure (such as bacterial infection), ASL in CF lungs can easily collapse quickly. The lungs of babies with CF are sterile at birth, but they will be in a state of continuous lung infection when they are one or two years old. The accumulation of mucin on the surface of the trachea and dehydration may be the first step of CF lung disease.
References
- Proesmans M, et al.; What's new in cystic fibrosis? From treating symptoms to correction of the basic defect. Eur J Pediatr. 2008;167(8):839-849.
- Randak CO, Welsh MJ. Role of CFTR's intrinsic adenylate kinase activity in gating of the Cl(-) channel. J Bioenerg Biomembr. 2007, 39(5-6):473-479.
- Smith JJ, et al.; Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996, 85(2):229-236.
- Ratjen F, et al.; Effect of inhaled tobramycin on early Pseudomonas aeruginosa colonisation in patients with cystic fibrosis. Lancet. 2001;358(9286):983-984.
- Cutting GR, Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015, 16(1):45-56.
Inquiry
