Dent's Disease
Dent disease is an X-linked recessive genetic disease. It was first reported by Dent et al. in 1964. The clinical features include low molecular proteinuria (LWMP), hypercalciuria, nephrocalcinosis, and progressive renal failure, among which 30 %-80% of patients develop to the end stage of kidney disease when they are 30-50 years old.
Dent Disease Genotype And Clinical Classification
Gene mutations are the cause of Dent's disease, about 60% of which are caused by voltage-gated chloride channel protein 5 (CLCN5) gene mutations, and 15% are caused by eye-brain-renal syndrome related gene 1 (OCRL1) gene Caused by mutations. CLCN5 is located on chromosome Xp11.23-p11.22 and consists of 12 exons, encoding Cl-channel protein-5 (CLC-5) consisting of 746 amino acids. The disease caused by CLCN5 gene mutation is called Dent disease type 1 At present, more than 200 gene mutations have been reported. OCRL1 is located on chromosome Xq24-26 and contains 5152 nucleotide base pairs and 24 exons, 23 of which encode the OCRL1 protein with phosphatase activity. OCRL1 mutation causes type 2 Dent disease. The known Dent disease patients are all caused by a single gene, but Addis et al. reported 1 case, which was caused by the CLCN5 gene and OCRL1 gene.
At present, there are still 25% of patients with no gene mutations detected, so there may be other pathogenic genes. Collectrin gene (also known as TMEM27) is located on chromosome Xp22.2, with a total length of 1262 bp, which encodes a transmembrane glycoprotein composed of 222 amino acids. This gene product is expressed in the apical membrane of the renal tubule and participates in the regulation of amino acid transport. Animal experiments show that the Collectrin gene can increase the expression of the B0AT1 transporter, thereby enhancing amino acid transport. Collectrin knockout mice developed obvious amino aciduria, and as the symptoms got worse, too much amino acid was lost, and crystaluria would also appear. Therefore, some scholars speculate that the TMEM27 gene may be one of the pathogenic genes of Dent disease, but so far no TMEM27 gene mutation has been detected in patients.
Pathogenesis
CLCN5 Gene Mutation
Studies have shown that CLC-5 is not entirely a Cl- channel, but a 2Cl-/H+ exchanger, but the specific relationship between the function of this exchanger and the etiology of Dent's disease is still unclear. Novarino et al. designed a knock-in (KI) mouse with a gated glutamate mutation. This mutation can convert CLC-5 from a 2Cl-/H+ exchanger to a pure Cl- channel. Comparing it with ordinary knockout (KO) mice, it is found that KI mice with normal endosome acidification will also have LWMP. This study confirmed that the concentration of Cl- in the endosome is an important guarantee for maintaining endocytosis. Under normal circumstances, the acidification of endosomes causes the dissociation of the Megalin/Cubilin multi-ligand complex, and Megalin/Cubilin is recycled and reused in the apical membrane, while the remaining ligands are directly degraded by lysosomes. Abnormal endosomal acidification caused by CLCN5 gene mutation can lead to abnormal dissociation of Megalin/Cubilin complex, impaired endocytosis, and increased loss of small molecules such as vitamin D binding protein, 25-hydroxyvitamin D3, and parathyroid hormone (PTH). Stéphane et al. divided the reported CLCN5 mutations into three types: 60% of the mutations occurred in the CLC-5 homodimer binding surface, presumably affecting the CLC-5 post-transcriptional processing and homodimer formation. Cause CLC-5 to stay in the endoplasmic reticulum and degrade prematurely; 20% of mutations occur on the edge of the homodimer surface, which is speculated to have little effect on the protein structure. CLC-5 is normally distributed in the early endocytosis and will not be retained In the endoplasmic reticulum, it will affect the stability of mature CLC-5 and reduce the accumulation on the surface of renal tubular epithelial cells; there is another type of mutation that does not affect the distribution of CLC-5 but changes 2Cl-/H+ to Cl-/H+, Although the acidification function of endosomes is not affected, it leads to changes in Cl-current. At present, the formation mechanism of hypercalciuria in patients with Dent disease is not clear. It is generally believed that the high concentration of PTH in urine stimulates the synthesis of vitamin D3, which increases the reabsorption of Ca2+ by the renal tubules and small intestine, the leakage of Ca2+ from the kidney, and the increase of calcium reabsorption by bones. There may be other mechanisms that cause Ca2+ to be lost from the kidney, but it is not yet fully understood. PTH reabsorbs in the proximal tubule decreased harvest, increased urinary PTH concentration, activated the apical membrane PTH receptor, and then activated the sodium-phosphorus cotransporter of the brush border, which reduced the reabsorption of phosphorus in the proximal tubules, resulting in hyperphosphateuria.
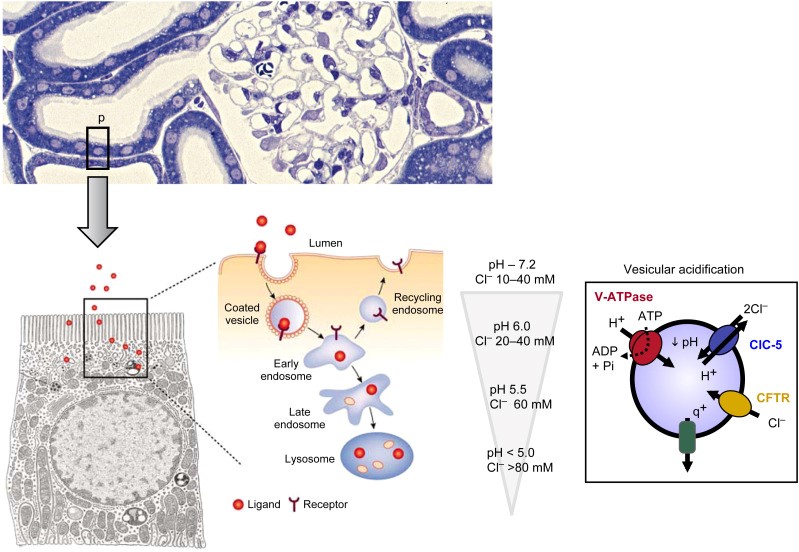
Figure 1. The molecular model of Dent disease. (OlivierDevuyst, et al. 2018)
OCRL1 Mutation
OCRL1 protein is a phosphatase produced by the OCRL1 gene, which can hydrolyze phosphatidylinositol 1,4,5-triphosphate to produce phosphatidylinositol 4,5-bisphosphate. In kidney epithelial cells, it is mainly distributed in lysosome and Golgi complex, and can also be distributed in endosome to participate in endocytosis. OCRL1 protein includes three main functional regions: the central inositol 5-phosphatase region, the RabGTPase region for ASH binding, and the catalytically inactive RhoGAP-like C-terminal region. The specific role of OCRL1 has not been thoroughly studied, but functional studies have shown that OCRL1 is involved in the transport process of the endosome and the cis-Golgi reticulum system. Recent studies have shown that OCRL can affect early endosome function through its 5-phosphatase activity. When OCRL is excessively depleted or expression is missing, it can reduce the recycling and reuse of Megalin and make it stay in the early endocytosis, thereby affecting the reabsorption of urine protein. Transient receptor potential vanillin subfamily 6 (TRPV6) is an intestinal epithelial Ca2+ channel that participates in the active reabsorption of Ca2+. Under normal circumstances, OCRL protein affects Ca2+ reabsorption by regulating the PI(4,5)P(2) required for TRPV6 to function. Studies have shown that after the Xenopus OCRL gene is knocked out, the activity of PI(4,5)P(2)5-phosphatase is reduced, and PI(4,5)P(2) is increased, thus making TRPV6-mediated intestinal tract The reabsorption of Ca2+ increases, the excretion of Ca2+ from the urine increases, and eventually hypercalciuria appears.
References
- Tosetto E, et al. Phenotypic and genetic heterogeneity in Dent's disease-the results of an Italian collaborative study. Nephrol Dial Transplant. 2006, 21(9):2452-2463.
- Malakauskas SM, et al. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene Collectrin. Am J Physiol Renal Physiol. 2007, 292(2): F533-544.
- Novarino G, et al. Endosomal chloride-proton exchange rather than chloride conducttance is crucial for renal endocytosis. Science, 2010, 328(5984): 1398-1401.
- Cui S, et al. OCRL1 function in renal epithelial membrane traffic. Am J Physiol Renal Physiol. 2010, 298(2): 335-345.
- OlivierDevuyst, et al. Genetics of Bone Biology and Skeletal Disease. Chapter 41 - Renal Fanconi Syndrome, Dent Disease, and Bartter Syndrome. 2018, https://doi.org/10.1016/B978-0-12-804182-6.00041-1
Inquiry
