Focal Segmental Glomerulosclerosis
Focal segmental glomerulosclerosis (FSGS) is a pathological diagnosis characterized by partial glomerular segmental scar formation and partial foot process disappearance. It is a common cause of nephrotic syndrome (NS) and accounts for most of adult cases. 40% and 20% of children, and is one of the main causes of hormone-resistant nephrotic syndrome (SRNS) and end-stage renal disease (ESRD), and the incidence has gradually increased in recent years. FSGS is mainly divided into primary FSGS and secondary FSGS. Primary FSGS is currently believed to be mainly due to the existence of some circulating permeability factors/cytokines, leading to the disappearance of podocyte foot processes and proteinuria. The main causes of secondary FSGS include genetic factors, viruses, drugs, diabetes, hypertension, obesity, renal hypoplasia, renal artery stenosis, cholesterol embolism, and vascular diseases. A variety of gene mutations have been found to be related to the occurrence and development of FSGS, and here is a brief review.
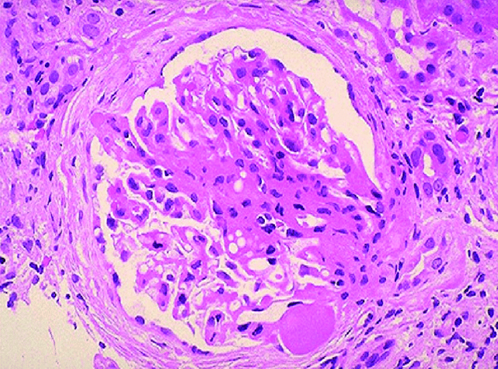
Figure 1. This is a photomicrograph of focal segmental glomerulosclerosis (FSGS).(Adanze Asinobi, et al.; 2015.)
Genes Mutations Related to Slit Diaphragm
1. Nephropathy protein 1 (NPHS1), nephropathy protein 2 (NPHS2)
The nephrin protein encoded by NPHS1 is an adhesion protein that is expressed at the junction between podocytes in the glomerulus. The mutation of human NPHS1 causes the development of the foot process structure to fail. Decreasing the expression of nephrin in mature glomeruli in animal models resulted in histological changes similar to FSGS. Podocytes with low levels of nephrin expression are both sensitive and difficult to repair from damage. NPHS1 gene mutations are mainly missense mutations. In addition to the Finnish congenital NS, which can cause autosomal recessive inheritance, it can also cause FSGS in children and adults. The podocin membrane protein encoded by NPHS2 is an important part of the slit diaphragm, and it interacts with nephrin protein and CD2 related protein (CD2AP). Mutations in NPHS2 can cause changes in podocin and thus change the filtration rate of the slit membrane. NPHS2 mutations are usually inherited recessively.
2. CD2AP
CD2AP is a podocyte split membrane molecule encoded by the CD2AP gene, which maintains the nodule of podocytes in the glomerulus. It plays an important role in structure and function. Takano et al. found homozygous frameshift mutations in CD2AP in families with FSGS and end-stage renal disease in childhood, and introduced the same frameshift mutations in mice through gene editing. Mice developed FSGS and renal failure, proving that homozygous mutations of CD2AP lead to human FSGS.
3. Transient receptor potential cation channel 6 (TRPC6)
TRPC6 is expressed in the glomeruli and tubules and is involved in a variety of chronic kidney diseases, including FS-GS and renal fibrosis after ureteral obstruction. It is a non-selective cation channel. It participates in podocyte expression and interacts with nephrin, podocin and other slit membrane molecules. Gheissari et al. tested the exons 2 and 13 of the TRPC6 gene in 26 Iranian FSGS children under 16 years of age and concluded that TRPC6 could be used for genetic screening of Iranian FSGS children. Zhang et al. conducted TRPC6 mutation screening on Chinese FSGS families and found that the TRPC6 mutation rate was 2.5%.
Podocyte Skeleton Related Molecules
Inverted formin2 (INF2) is one of the main autosomal dominant pathogenic genes that can cause FSGS. INF2 protein can directly remodel the actin filaments in vitro, and can also regulate intracellular actin dynamics and actin-dependent cellular behavior by inhibiting Rhoa/Dia signals. Because INF2 mutations disrupt the ability of INF2 to regulate Rho/Dia-mediated actin dynamics, individuals with INF2 mutations often have moderate proteinuria during adolescence or early adulthood, and both disease and proteinuria are progressive. Often leads to ESRD. Subramanian et al. found that mouse glomerular development does not require normal INF2 function, but the actin-based glomerular response to injury and repair behavior requires normal INF2 regulation. Recent studies have also shown that INF2 may be related to kidney histology other than focal segmental glomerulosclerosis. The genetic spectrum of INF2-related diseases may be broader. In addition to INF2, α-actinin 4, cyclin actin binding protein, Rho GDP dissociation inhibitor α, RhoGTPase activator protein 24, and tetrameric peptide protein 21B gene mutations can also cause changes in the podocyte cytoskeleton, leading to FSGS. Some of these related proteins are part of the podocyte cytoskeleton, and some can stabilize and repair podocytes. Changes in their proteins may cause damage to podocyte movement dynamics. But these mutations are relatively rare and there are few clinical reports.
Podocyte Transcription Factor
1. LIM Homeobox Transcription Factor 1-beta (LMX1B)
LMX1B is related to podocyte dysfunction. Its protein is synthesized in podocytes and plays a key role in glomerular development. The increase in glomerular volume of Lmx1b knockout mice significantly decreases and glomerular sclerosis increases.
2. Paired Box Gene 2 (PAX2)
PAX2 is a transcription factor involved in kidney development and phenotypic regulation of glomerular epithelial cells. In one study, PAX2 mutations accounted for a significant proportion of families with hereditary FSGS (about 4%). PAX2 mutations can also cause congenital abnormalities in the kidneys and urinary tract. In vitro studies have shown that several PAX2 mutations related to FSGS interfere with protein function by affecting the correct binding and transcription of DNA or by changing the interaction between PAX2 and repressor protein.
3. Wilms Tumor 1 (WT1)
Various mutations in WT1 have been identified as the cause of syndromes of hereditary FSGS and diffuse mesangial sclerosis. The zinc finger DNA binding protein encoded by WT1 is essential for kidney, urinary tract and gonadal development. The phenotype of the WT1 mutation associated with FSGS is mainly Frasier syndrome. Mutations in the carboxy-terminal zinc finger domain encoded by exons 8 and 9 of WT1 impaired the transcriptional activation function of podocyte-specific nuclear transcription factors, resulting in significant gene expression of basic slit diaphragm components such as nephrin, podocin, and podocyte marker proteins change.
In addition, studies have found mitochondrial-related gene mutations, such as aarF domain containing kinase 4, coenzyme Q6 homologue, and coenzyme Q2 homologs, decaprenyl diphosphate synthase subunit, and collagen type IV alpha 3, 4, 5, all cause FSGS.
References
- Adanze Asinobi, et al.; Trends in the histopathology of childhood nephrotic syndrome in Ibadan Nigeria: Preponderance of idiopathic focal segmental glomerulosclerosis. BMC Nephrology. 2015, DOI: 10.1186/s12882-015-0208-0.
- Rakesh V, et al.; Nephrin is necessary for podocyte recovery following injury in an adult mature glomerulus. PLoS One. 2018, 13(6): e0198013.
- Huber Tobias B, et al.; Podocin organizes ion channel -lipid supercomplexes:implications for mechanosensation at the slit diaphragm. Nephron Exp Nephrol. 2007, 106(2): e27-e31.
- Brown EJ, et al.; Mutations in the formingene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010, 42(1): 72-76.
- Kaltenis P, et al. Slow progressive FSGS associated with an F392L WT1 mutation. Pediatr Nephrol. 2004, 19(3): 353-356.
Inquiry
