Hyperkalemic Periodic Paralysis
Hyperkalemic periodic paralysis (HYPP) is an autosomal dominant genetic disease. It affects sodium channels in muscle cells and affects the level of potassium particles in the blood. The disease is characterized by excessive excitement or weakness of the muscles, which is exacerbated by excessive potassium intake and heat or cold stimulation, which can lead to uncontrolled shaking, which can lead to paralysis. Onset usually occurs in early childhood, but it also occurs in adults. Depending on the type and severity of HYPP, the disease may increase or stabilize. After 40 or 50 years, the onset may decrease or stop, or it may continue to develop into old age depending on the type. The patient's muscle strength usually increases during an attack, although as the disease progresses, many affected people may experience increasing muscle weakness. In addition, patients with HYPP may experience some degree of muscle stiffness and cramps (muscle rigidity). This may be caused by the same factors that trigger the paralysis, depending on the type of myotonic muscle.
Between episodes of hyperkalemic periodic paralysis, the patient's blood K+ is often higher than normal, and the K+ concentration changes after meals; myalgia and/or muscle rigidity may be present during the attack, and the serum K+ rises. Rest after exercise, oral potassium, cold, pregnancy, and intake of mineralocorticoids often induce muscle weakness. Serum K+ rises during exercise; drops after stopping, almost close to the initial level. It rose again after 20-30 minutes, reaching the maximum value of 7mM in about an hour, and fell again within the second hour, often lower than normal. During the second blood K+ rise, paralysis occurred. The blood K+ concentration measured by arteries and veins showed that the second increase in blood K+ came from the outflow of K in the muscle, which caused the decrease of K+ in the muscle. Part of the extra plasma K+ is excreted through the kidneys. After the onset, when the muscles re-uptake K+, low blood K+ appears. The study of hyperkalemic periodic paralysis of the intercostal muscles with and without myotonia found that in a solution with K+ of 3.5 mM, the resting potential is normal; increase K+ to 7 mM, and the muscle fibers are depolarized. The excitability increased to produce repeated electrical activity, and the repeated electrical activity disappeared, and then paralysis.
In humans, the most common underlying genetic cause of hyperkalemic periodic paralysis is a point mutation in the gene SCN4A. This gene encodes the voltage-gated sodium channel Nav 1.4 at the neuromuscular junction. Since the disease is inherited in an autosomal dominant manner, even one copy of the altered gene in each cell will trigger HYPP.
Under normal physiological conditions, action potentials from the central nervous system cause endplate potentials at the neuromuscular junction, which causes sodium ions to enter Na v 1.4 and depolarize muscle cells. This depolarization triggers the entry of calcium from the sarcoplasmic reticulum, causing muscle contraction (tightening). To prevent permanent muscle contraction, the channel includes a quick-kill door that blocks it quickly after opening. This prevents further entry of sodium. Over time, potassium ions leave the muscle cells, repolarizing the cells, and causing calcium ions to be pumped out of the contraction device, thereby relaxing the muscles.
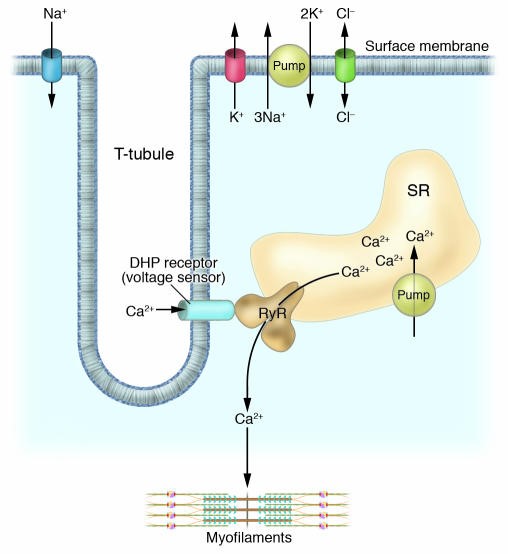
Figure 1.Excitation-contraction coupling of skeletal muscle. (Karin Jurkat-Rott, et al. 2005)
Therefore, mutations that change the general structure and function of the sodium channel can disrupt the regulation of muscle contraction, resulting in severe muscle weakness or paralysis. Mutations have been identified in residues between Nav 1.4 transmembrane domains III and IV, and these residues constitute the rapid inactivation gate of Nav 1.4. There are also mutations in the cytoplasmic loop between the S4 and S5 helices of the II, III and IV domains, which are the binding sites of the inactivation gate. Therefore, these mutations can potentially cause hyperkalemic periodic paralysis.
The pathological mechanism of SCN4A mutation in hyperkalemic periodic paralysis is very complicated, but it can explain the hyperkalemia correlation of the disease. In patients with SCN4A mutations, not all channel copies are inactivated after the action potential. This causes sodium to leak and cannot return to the original resting membrane potential. Under hyperkalemia conditions, this causes additional chronic depolarization of the membrane potential and sodium leakage. This sodium leakage raises the potential of all sodium channels and releases them from inactivation (into the depolarization module). Since the motor endplate has been depolarized, further contraction signals are invalid, and ultimately lead to paralysis.
References
- Rojas CV, et al.; A Met-to-Val mutation in the skeletal muscle Na+ channel alpha-subunit in hyperkalaemic periodic paralysis. Nature. 1991, 354 (6352): 387-9.
- Bendahhou S, et al.; Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology. 2002, 58 (8): 1266-72.
- Jurkat-Rott K, Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J. Clin. Invest. 2005, 115 (8): 2000-9.
- Karin Jurkat-Rott, et al. Muscle channelopathies and critical points in functional and genetic studies. J Clin Invest. 2005, 115(8): 2000-2009.
Inquiry
