Hypokalemic Periodic Paralysis
When the genes encoding ion channel subunits undergo mutations/expression abnormalities, or pathological endogenous substances appear in the body for the channels, the function of the channels is reduced or enhanced to varying degrees, which leads to the disorder of the body's overall physiological functions, leading to certain Some congenital and acquired diseases, this type of disease is called "channelopathy". Since the 1990s, ion channel diseases have become a research hotspot in the medical and biological circles. In the nervous system, it is mainly divided into central nervous system ion channel diseases and skeletal muscle ion channel diseases. Since 1994, when Ptacek et al. discovered that Familial Hypokalemic Periodic Palsy (FHypoPP) had mutations in genes encoding Ca2+ ion channels, FHypoPP has become the most common clinical scientist recognized skeletal muscle ion channel disease. With the widespread development of patch clamp and gene cloning and sequencing technologies, people have gradually deepened the research on the genetic etiology and pathogenesis of hypokalemic periodic paralysis.
Primary hypokalemic periodic paralysis
FHypoPP
FHypoPP is inherited in an autosomal dominant manner, and the penetrance rate in women is low, and it tends to occur in young adults. It can be induced by injections of insulin, corticosteroids or large amounts of glucose, as well as strenuous exercise, full meals, emotional agitation, menstruation and infection trauma. The typical manifestations are paroxysmal limbs paralysis of the extremities, decreased muscle tone, and weakened or disappeared tendon reflexes. Recent studies have found that 69% of FHypoPP-related genes are voltage-gated calcium channel CACNA1S genes, 8.6% are voltage-gated sodium channel SCN4A genes, and the remaining 22.4% are still unknown.In addition, a small portion patients has KCNE3 genes mutation which encode potassium channels. Scientists divide the FHypoPP corresponding to the above three ion channel changes into three types: FHypoPP-1, FHypoPP-2, FHypoPP-3. FHypoPP-1: In 1994, Ptacek et al. first discovered a genetic defect in chromosome 1q31-32 in a case of FHypoPP, which was specifically located in CACNA1S. This gene encodes the muscle dihydropyridine (DHP) sensitive Ca2+ channel α1 subunit. The following allelic variants were found: Arg1239His, Arg1239Gly, Arg528His, and speculated that this mutation interferes with depolarization signal transmission, weakens the excitation-contraction coupling, and causes disease. Some also believed that the depolarization caused by hypokalemia reduces the release of calcium ions, directly or indirectly affecting the voltage gate control of sodium channel inactivation, so that the muscle fiber membrane cannot be excited and muscle paralysis occurs. It may also be related to the Na+ -K+ -ATPase. FHypoPP-2: Cllinical studies find a mutation (Arg669His) of the SCN4A gene encoding the alpha subunit of the voltage-gated sodium channel appeared in some cases, which was located on chromosome 17q22-23. Struyk pointed out that the sodium channel with R669H mutation exhibited a hyperpolarization movement of about 10 mV during slow inactivation, which prolonged the recovery period by two to five times after depolarization. Therefore, it can be judged that the mutation may have no obvious effect on the rapid inactivation, but it will promote the slow inactivation of sodium channels. In 2000, Jurkat-Rott proposed three new mutation sites of Arg672His, Arg672Gly and Arg672Ser. The pathogenesis may be that the sodium channel defect caused by the mutation strengthens the rapid inactivation of the sodium channel and also affects the slow inactivation process. . FHypoPP-3: In familial hypokalemic periodic paralysis, the voltage-dependent potassium channel gene KCNE3 is defective in the type called FHypoPP-3, which accounts for 2% of the total number of diseases. This gene encodes the polypeptide MiRP2, and MiRP2 and its subunit Kv3.4 form a MiRP2-Kv3.4 complex, which is the A-type channel of skeletal muscle. It is more powerful than ordinary channels, because it changes the conductivity, opening probability and sensitivity to peptide toxins of a single channel, allowing more K+ to pass, and accelerating the recovery process of activation. So far, people have found only one mutation site of this gene: Arg83His. Studies have shown that the abnormal complex MiRP2-Kv3.4 formed by this mutation reduces the outward current that forms the action potential, reduces the ability to set the resting potential, and turns the resting potential to an abnormal depolarization voltage, which is a the direct cause for muscle dysfunction.
Sporadic Hypokalemic Periodic Paralysis (SHypoPP)
The clinical features of SHypoPP are similar to those of FHypoPP, except that the age of onset is slightly older, and SHypoPP does not have frequent and severe attacks. The study found that only some cases of SHypoPP have the same genetic mutation as FHypoPP.
Anderson Syndrome (Andersen-Tawil syndrome)
In 1971, Andersen et al. first called Anderson syndrome another type of primary periodic paralysis. The disease is inherited in an autosomal dominant manner, with characteristics of phenotypic variation within the family, incomplete penetrance, and frequent mutations. All patients are accompanied by congenital developmental abnormalities, such as shortness, low ear drops, small mandible, wide nose, digit-to-palatine deformities, etc.; 80% of patients have prolonged QT interval and ventricular arrhythmia; muscle weakness may be accompanied by hypokalemia (about 78%), normal blood potassium or hyperkalemia. The first mutation gene that can cause this disease is the KCNJ2 gene located on chromosome 17q, which encodes a voltage-independent potassium channel. Up to now, about 20 mutations have been identified, such as D71V, S136F, G144S and so on. Since Andersen's family coefficient is very small, no clear connection between genotype and phenotype has been found. It is believed that the KCNJ2 mutation interferes with the normal excitement of cells, causes polarization recovery defects, and causes abnormal performance of myocardium and skeletal muscle. The characteristics of deformity may be attributed to a direct effect on tissue migration or development.
Secondary Hypokalemic Periodic Paralysis
There are many types of secondary HypoPP, such as secondary to primary aldosteronism, renal tubular acidosis, diabetic acidosis, scleroderma etc.. Among the secondary periodic paralysis, the most common and most important one is thyrotoxic hypokalemic periodic paralysis (THypoPP).
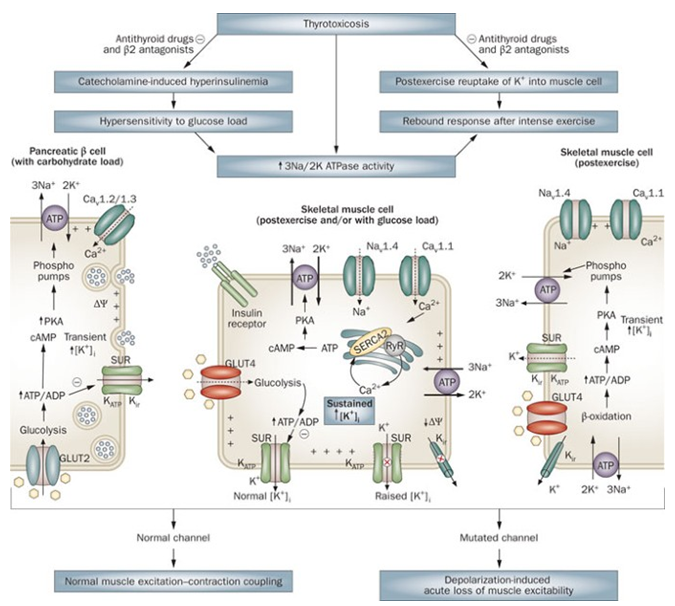
Figure 1. Thyrotoxic periodic paralysis pathophysiology hypothesis.(Rui M. B. Maciel, et al.; 2011)
More than 95% of patients with THypoPP are sporadic cases, predominantly between 20-40 years old. High carbohydrate intake, hyperinsulinemia and exercise can induce this disease. Its clinical manifestations are mostly weak proximal muscles, which can also involve the respiratory muscles and the innervating muscles of the medulla oblongata, causing serious consequences such as dyspnea. Attacks often last several hours to several days, with or without symptoms of hyperthyroidism, and there is no correlation between attacks and the severity of hyperthyroidism. On the basis of active treatment of hyperthyroidism, supplemental KCl treatment is effective in restoring muscle strength. Cerutti et al. found that the potassium channel gene KCNE3 has an Arg83His mutation. It is proposed that the pathogenesis of ThypoPP may be the same as that of FHypoPP-3, that is, the mutation reduces the inflow of external K+, forms a more active resting membrane potential, and reduces repolarization ability. Subsequently, the sodium channel SCN4A gene Arg672Ser mutation was also confirmed. So far, people have not found the same Ca2+ channel gene mutation as FHypoPP. Some people believe that the voltage-dependent Ca2+ channel gene single nucleotide polymorphism is related to the pathogenesis of THypoPP, and the loss of gene polymorphism will change the effect of thyroxine on the Ca2+ channel, thereby causing patients to become sick. In addition, it is speculated that the enhancement of Na+ /K+ pump activity is also one of the pathophysiological basis of K+ influx into cells and low potassium. Studies by many groups have shown that the number of Na+ /K+ pumps of white blood cells and platelets in THypoPP patients, as well as the influx of cations involved in Na+ /K+ -ATPase, have increased. In addition, thyroxine can enhance the excitatory effect of adrenaline on Na+ /K+ -ATPase and increase the responsiveness of Na+ /K+ pump to insulin. Therefore, most patients have hyperinsulinemia.
References
- Ptacek LJ, et al.; Dilhydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. 1994, 77(6): 863-868.
- Struyk AF, et al.; The human skeletal muscle Na + channel mutation R669H associated with hypokalemic periodic paralysis enhanced slow inactivation. J Neurosci. 2000, 20(23): 8610-8617.
- Kung AW, et al.; Association of novel single nucleotide polymorphisms in the calcium channel alpha 1 subunit gene (Cav1.1) and thyrotoxic periodic paralysis. J Clin Endocrinol Metab. 2004, 89(3): 1340-1345.
- Bendahhou S, et al.; Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. The journal of biological chemistry. 2003, 278(51): 51779-51785.
- Rui M. B. Maciel, et al.; Novel etiopathophysiological aspects of thyrotoxic periodic paralysis. Nature Reviews Endocrinology. 2011, volume 7, pages657-667.
Inquiry
