Juvenile Myoclonic Epilepsy
Juvenile myoclonic epilepsy (JME), also known as Janz syndrome, was included in the international classification of epilepsy and epilepsy syndrome in 1989. JME is one of the most common idiopathic (hereditary) and general epilepsy syndromes (IGEs), accounting for 5% to 10% of all epilepsy patients, and the incidence of women is higher than that of men. JME begins around puberty, and the age of onset is usually 12 to 18 years old. The clinical manifestations are the triad of myoclonus, absence, and generalized tonic-clonic seizures. The electroencephalogram during the seizure period shows bilateral synchronous and symmetrical multi-spinous slow complex wave bursts, and the interictal period is generally irregular 3-6 Hz Spine slow or multi-spine slow complex waves are emitted. Genes related to JME currently reported include voltage-gated calcium channel-related genes (CACNB4), GABA receptor-related genes (GABRA1, GABRD), voltage-gated chloride channel-related genes (CLCN2), and non-ion channels Class-related genes (EFHC1) and other related genes (CASR, BRD2).
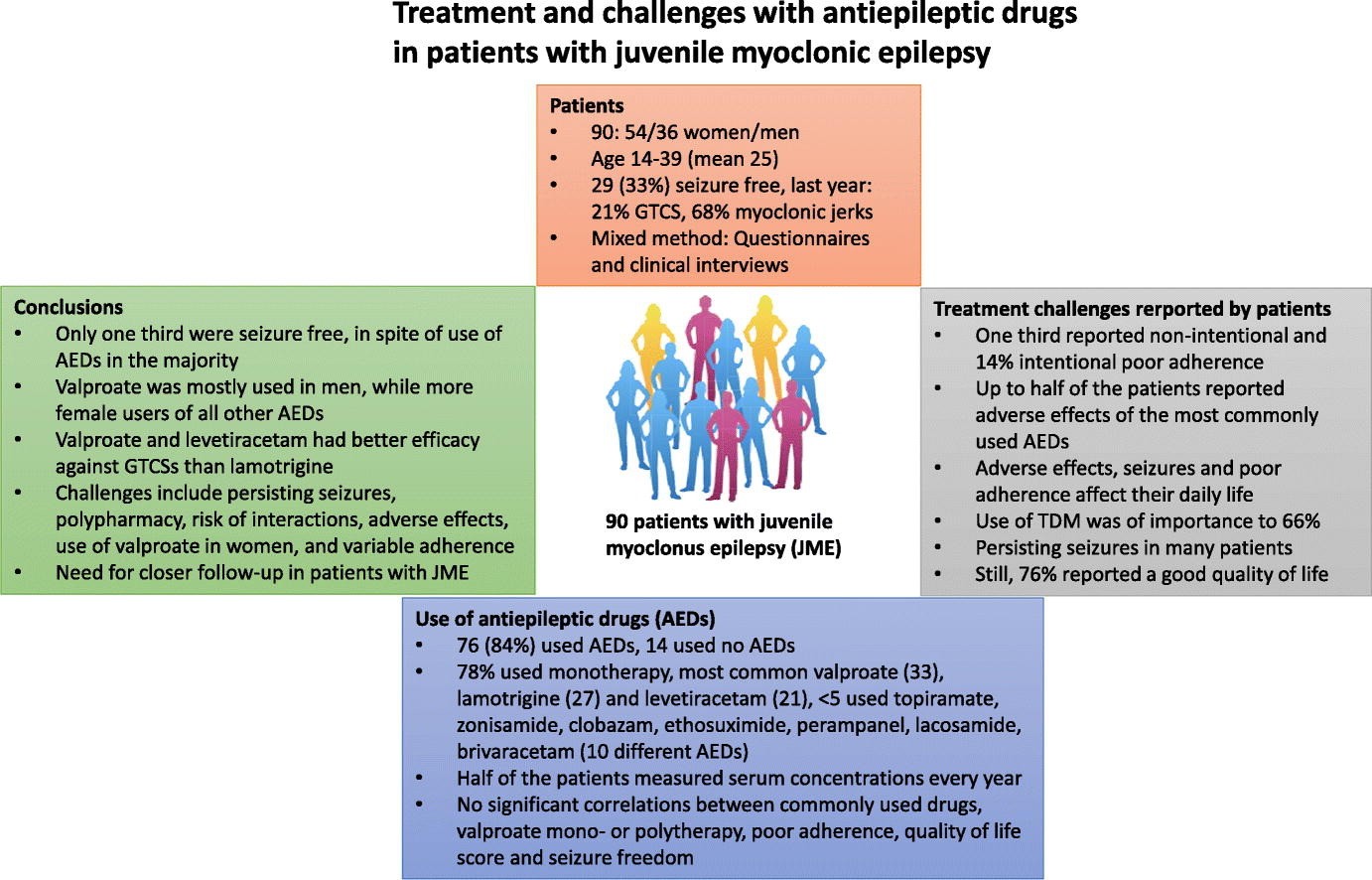
Figure 1.Treatment and challenges with antiepileptic drugs in patients with juvenile myoclonic epilepsy(Johannessen Landmark C, et al., 2019)
Calcium Channel Related Gene (CACNB4)
CACNB4 is a gene encoding the β4 subunit of a voltage-gated calcium ion channel located at 2q22~23. It plays an important role in regulating calcium ion flow and calcium ion channel activation and inactivation. It is widely distributed in the cerebellum. The calcium ion channel is essentially a transmembrane glycoprotein that forms a funnel-shaped hydrophilic pore, which acts as a selective valve for ions, and plays an important role in the generation and transmission of nerve signals and the regulation of cell excitability. At present, it is believed that a short and rapid calcium ion influx causes cell depolarization. This depolarization will trigger sodium ion influx to a certain extent, and a series of rapid depolarization processes will erupt, eventually leading to abnormal discharge of brain neurons.
There are two β4 subunit mutations associated with juvenile myoclonic epilepsy, namely C104F and R482X. Escayg et al. found that voltage-gated calcium channels containing R482X mutations expressed on oocytes can generate 30%-40% more current than wild-type, and the inactivation rate is also faster than wild-type. The C104X mutation causes the deletion of 38 amino acids at the C'end of the related amino acid chain, causing the loss of important functional domains in the voltage-gated calcium channel, resulting in abnormal calcium channel function, and ultimately causing abnormal discharge in the brain. In 2012, Tadmouri and other studies found that the mutant β4 subunit can affect the coupled phosphatase at its specific site, thereby affecting the transmission of nerve signals, and eventually leading to seizures.
GABA Receptor-Related Genes
GABRD is a gene encoding the δ subunit of the GABA receptor, located at 1p36.33. There are 3 mutations related to JME, namely E177A, R220C, and R220H. Dibbens et al. found that the GABA receptor containing the E177A mutation significantly reduced the amount of current passed by the wild-type receptor, and the amplitude was significantly lower, and it was more serious than the R220C and R220H mutations. Concurrent studies have shown that receptors containing E177A mutations are not only less responsive to GABA than wild-type receptors, but also have a significantly reduced expression on the cell membrane.
Chloride Channel Related Gene (CLCN2)
CLCN2 is a gene encoding a voltage-gated chloride channel, located at 3q27.1, and is highly expressed in GABA-suppressed brain regions. Haug et al. identified three CLCN2 mutations in three IGEs families: the first is an immature stop codon mutation, the second is a splicing mutation, and the third is a missense mutation G715E. And proposed that the CLCN2 mutation is related to Juvenile myoclonic epilepsy (JME), juvenile absence epilepsy (JAE), and childhood absence epilepsy (CAE) are related. In 2004, Niemeyer et al. studied the effects of CLCN2 in human embryonic kidney cells transfected in vitro through patch clamp and confocal microscopy. They found that termination mutations and splicing mutations can lead to the loss of chloride channel function and reduce the chloride ion concentration gradient across the membrane, which is important for suppresses function of GABA. The G715E mutation can change the voltage gating, which may lead to depolarization and hyperexcitability of nerve cells.
In addition, study also found that mutations in the EFHC1 gene that encodes the EF hand-shaped domain that binds to calcium, the BRD2 gene that encodes nuclear transcription regulators, and the CASR gene that encodes calcium-sensitive receptors can also cause JME.
References
- Camfield CS, et al. Epidemiology of juvenile myoclonic epilepsy. Epilepsy & Behavior. 2013, 28(7): S15-S17.
- Dibbens LM, et al.GABRD encoding a protein for extra or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Human Molecular Genetics. 2004, 13(13): 1315-1319.
- Feng HJ, et al. Delta subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of alpha4 beta2 delta GABAA receptors.Journal of Neuroscience the Official Journal of the Society for Neuroscience. 2006, 26(5): 1499-1506.
- Johannessen Landmark C, et al. Treatment and challenges with antiepileptic drugs in patients with juvenile myoclonic epilepsy. Epilepsy Behav. 2019, 98(Pt A):110-116.
Inquiry
