Malignant Hyperthermia
Malignant hyperthermia (MH) is a rare, autosomal-linked genetic disease of the muscular system. It is induced by the commonly used halogenated inhalation anesthetics and depolarizing muscle relaxants (succinylcholine). The patients of receiving general anesthesia has the incidence rate is 1/5000 to 1/1000. The ratio of male to female morbidity is 2.5 to 4.5.
Pathogenesis
It is currently recognized that MH is a metabolic disease of the muscular system. The main mechanism is that the concentration of Ca2+ in the skeletal muscle cytoplasm is uncontrolledly increased by the specific drugs, which triggers the continuous tonic contraction of muscle fibers, and subsequently a large increase in heat production and tissue hypoxia, acidosis, muscle cell necrosis, diffuse intravascular coagulation, and cardiovascular breakdown. The Ca2+ in the cytoplasm mainly comes from the sarcoplasmic reticulum, which is caused by the release of a large amount of Ca2+ from the sarcoplasmic reticulum due to ion channel defects. In addition, the extracellular Ca2+ influx may also be involved in the onset of MH. Studies have confirmed that the level of Ca2+ in the skeletal muscle cytoplasm of patients with MH during the non-onset stage of the disease is also higher than that of normal people, which may imply that patients with MH may also have a higher background flow of Ca2+ across the membrane under normal conditions.
Molecular Mechanism
Several types of macromolecules have been found to be related to the development of MH. Among them, ryanodine receptor 1 (RYR1) is the most important. RYR receptors are Ca2+ release channels, which are roughly divided into types 1, 2, and 3, located in skeletal muscle, myocardium, and brain tissue, respectively. In patients with MH, due to RYR1 receptor function defects, the channel continues to open under the trigger of sensitive drugs. A large amount of Ca2+ flows out from the sarcoplasmic reticulum, which far exceeds the recovery capacity of the Ca2+ pump on the sarcoplasmic reticulum, resulting in continued strong muscle fiber contraction. Another type of macromolecular substance related to the onset of MH is the dihydropyridine Ca2+ channel (DHPR). DHPR is located on the membrane of the transverse tube of muscle cells. It is a voltage-gated channel and is closely related to RYR1 in space and function. When the sarcoplasmic membrane is depolarized, the transmembrane potential is rapidly transmitted to the transverse tube membrane, excites DHPR and finally causes the RYR1 receptor to open, and Ca2+ flows out from the sarcoplasmic reticulum. It is currently believed that 50% to 70% of MH is mediated by RYR1 and DHPR receptors. In addition, the muscle-type nicotinic acetylcholine receptor (nAChR) is thought to be related to the onset of MH triggered by succinylcholine. nAChR is a multimeric ligand-gated cation channel protein composed of 5 subunits, allowing K+, Na+, Ca2+ and other cations to pass. In existing literature reports, cases of MH induced by succinylcholine alone are very rare. In more cases, it may act by exciting nAChR and lowering the threshold of MH.
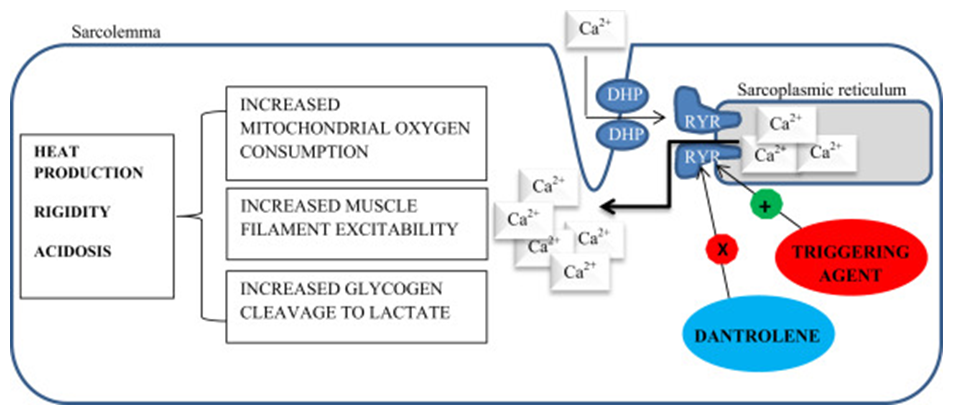
Figure 1. Malignant Hyperthermia.(Brian Butala, et al. 2018)
Genetic Mechanism
Abnormal function of receptors or ion channels is mainly controlled by genetic mutations. At present, the more certain genes related to the onset of MH mainly include the RYR1 and CACNA1S genes.
RYR1 Gene
RYR1 gene is located in 19q13.1-13.2, and 160 exons are encoded by 16000 base pairs. RYR1 gene mutations are very common. Since the first discovery of the relationship between RYR1 gene mutations and the onset of MH, more than 300 mutation sites have been confirmed, of which about 50 are related to the onset of MH. Most of the mutation sites are located in the MH/CCD 1, 2, and 3 regions (the so-called hot spots of malignant hyperthermia/central axis disease). A multi-center study of 200 cases showed that RYR1 mutations were detected in more than half of MH cases.
CACNA1S Gene
CACNA1S gene is located at 1q32, which encodes 44 exons by 93,500 base pairs and determines the amino acid sequence of the α1 subunit of DHPR. Gene mutations related to the onset of MH caused the 1086th amino acid residue on the α1 subunit to be changed from arginine to histidine.
In addition, studies have found that knocking out the CASQ1 gene encoding the endoplasmic reticulum in mice can induce symptoms similar to MH. However, there is currently no evidence that the CASQ1 gene is related to the onset of human MH.
References
- Monnicr N, et al. Prescnce of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. An-esthesiology. 2002,97(5):1067-1074.
- Eltit JM, et al.Nonspecific sarcolemmal cation channels are critical for the pat hogenesis of malignant hyperthermia.FASEBJ. 2013,27(3):991-1000.
- Yang T, et al.Enhanced excitation-coupled calcium entry in myotubes is associated with ex-pression of RyRl malignant hyperthermia mutations.J Biol Chem. 2007,282(52).37471-37478.
- Brian Butala, et al. Malignant Hyperthermia: Review of Diagnosis and Treatment during Cardiac Surgery with Cardiopulmonary Bypass. Journal of Cardiothoracic and Vascular Anesthesia. 2018, 32(6): 2771-2779.
Inquiry
