Mucolipidosis Type IV
Mucolipidosis type IV disease is a genetic disease characterized by developmental delay and visual impairment, and symptoms will worsen over time. The severe form of this disease is called typical mucolipidosis type IV disease, while the mild form is called mucolipidosis type IV disease.
Approximately 95% of people suffering from this disease have a severe form. People with typical mucolipidosis type IV have retarded development of mental and motor skills (including sitting, standing, walking, grasping objects, and writing) (psychomotor retardation). The psychomotor delay is form moderate to severe and usually becomes apparent in the first year of life. Affected people have intellectual disability, limited or lack of language skills, difficulty chewing and swallowing, weakened muscle tone (hypotonic) gradually becoming abnormal muscle stiffness (spasm) and problems with controlling hand movements. Most people with type mucolipidosis type IV disease cannot walk independently. In approximately 15% of affected individuals, psychomotor problems will worsen over time. In addition, people with typical type IV mucosal seborrheic disease may have normal vision at birth, but vision during the first decade of life got weaker and weaker. People with this disease will have the transparent covering of the eye (cornea) becoming cloudy, and the light-sensitive layer (retina) at the back of the eye will gradually decompose. By the time they reach their teens, the affected person will have severe vision loss or blindness.
The production of gastric acid (acid deficiency) is also impaired in people with typical mucolipidosis type IV disease. Although achlorhydria does not cause any symptoms in these individuals, it can cause abnormally elevated levels of gastrin (a hormone that regulates the production of gastric acid) in the blood. In addition, patients with type IV mucosal seborrheic disease may not have enough iron in the blood, which may lead to insufficient red blood cells (anemia). People with severe this disease can usually survive into adulthood. However, their life span may be shortened.
Approximately 5% of affected individuals have atypical mucolipidosis type IV. These people usually have mild psychomotor delays and may have the ability to walk independently. Patients with atypical mucolipidosis type IV have milder ocular abnormalities than severe patients. Achlorhydria may also be present in mild patients.
Pathogenesis
Studies have found that mutations in the MCOLN1 gene can cause type IV mucosal steatosis. This gene encodes the non-selective cation channel mucolipin1. The encoded protein is usually located in the membranes of lysosomes and endosomes, which are compartments for digestion and recovery of substances within the cell.
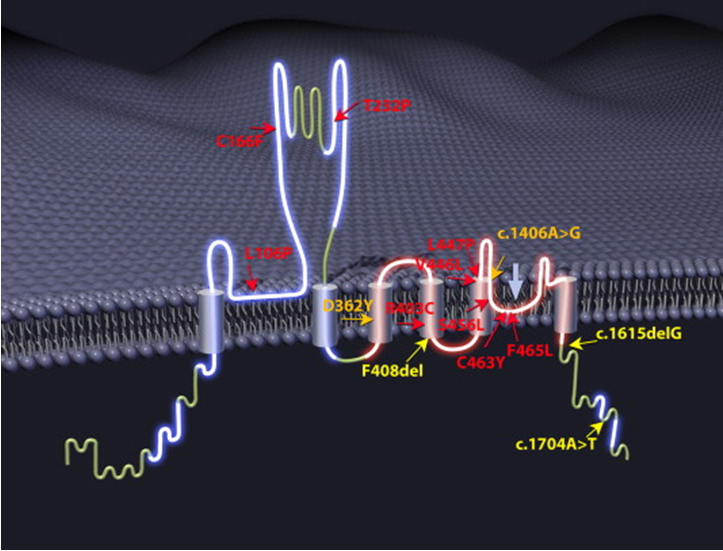
Figure 1. Mutations in MCOLN1. (Ehud Goldin, et al. 2015)
Mucolipin1 has a lower pH (acidification) in the endosome leading to protein degradation. There are at least 29 known MCOLN1 mutations in the entire gene. Many known mutations cause mucolipin1 not to be expressed. Lighter mutations can produce dysfunctional cation channels. Only mutations that change the C-terminus of the protein can also lead to a mild phenotype of the disease.
Although its function is not fully understood, mucin-1 plays a role in the transport of fats (lipids) and proteins between lysosomes and endosomes. Therefore, the lack of functional mucin-1 will impair the transport of lipids and proteins, leading to the accumulation of these substances in the lysosome, which in turn leads to diseases in which molecules accumulate in the lysosome, including type IV mucosal lipid disease. Mucolipin-1 seems to be important for the development and maintenance of the brain and retina. In addition, this protein may be essential for the proper functioning of cells in the stomach that produce digestive acid.
References
- Altarescu G, et al.; The neurogenetics of mucolipidosis type IV. Neurology. 2002, 59(3):306-13.
- Dong XP, et al.; The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008, 455(7215):992-6.
- Miedel MT, et al.; Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008, 205(6):1477-90.
- Puertollano R, et al.; TRPMLs: in sickness and in health. Am J Physiol Renal Physiol. 2009, 296(6):F1245-54.
- Ehud Goldin, et al. TRP Channels As Therapeutic Targets. Academic Press. 2015, ISBN: 978-0-12-420024-1.
Inquiry
