Myotonia Congenita
Myotonic myopathies (MM) is a group of primary/secondary ion channel proteins (Cl-/Na+/Ca2+/K+) structure/function abnormalities, skeletal muscle cell membrane depolarization disorder, and hereditary skeletal muscle diseases with continuous repetitive discharge. Clinical Manifestations are: muscle stiffness/myotonia, muscle atrophy/hypertrophy, muscle weakness; electrophysiology shows typical myotonia potential, with/without myopathy potential. According to clinical symptoms, it is divided into Dystrophic myotonia (DM) and Non-dystrophic myotonias (NDMs).
Non-Myotonic dystrophies (NDMs) are a group of hereditary skeletal muscle diseases characterized by a group of ion channel protein subunit coding gene mutations, increased excitability of skeletal muscle cell membranes and continuous repetitive discharge. The clinical manifestations include muscle rigidity, full muscle volume, muscle weakness (continuous/periodical attacks), fatigue, which may be accompanied by pain, occasionally involving heart and respiratory functions. According to the pathogenic gene, coding protein, and clinical phenotype, it is divided into Myotonia congenital (MC), Paramyotonia congenital, (PC), Hyperkalemic periodic paralysis (Hyper PP), Potassium-aggravated myotonia (PAM), and Hypokalemic periodic paralysis (Hypo PP) myotonia.
Among them, congenital myotonia is a disease that affects skeletal muscle movement. From childhood, people with this disease will experience constant muscle tension (muscle rigidity) that prevents normal muscle relaxation. Although myotonia can occur on any skeletal muscle, including the muscles of the face and tongue, it usually occurs in the legs. Congenital myotonia can cause muscle stiffness, which can interfere with normal movement. In some patients, the symptoms are very mild, and in severe cases, the symptoms may severely interfere with walking, running, and other activities of daily living. These muscle problems are especially obvious during the rest interval of exercise. Many affected patients find that repeated exercise can temporarily reduce their muscle stiffness, a phenomenon known as the warm-up effect.
The two main types of congenital myotonia are called Thomson's disease and Becker's disease. This is classified according to the severity of the symptoms of the disease and the way it is inherited. Becker's disease usually appears later in childhood than Thomson's disease and can cause more severe muscle stiffness, especially in men. People with Becker's disease often experience brief episodes of muscle weakness, especially in the arms and hands, caused by exercise after a period of rest. Over time, they will also experience mild permanent muscle weakness. This muscle weakness is not seen in patients with Thomson's disease.
Pathogenesis
Existing studies have shown that CLCN1 gene mutations cause congenital myotonia. Further research found that the CLCN1 gene directs the synthesis of related proteins that maintain the normal function of skeletal muscle cells. In order for the body to move normally, skeletal muscles must be tense (contracted) and relaxed in a coordinated manner. The contraction and relaxation of muscles are controlled by the flow of charged atoms (ions) into and out of muscle cells. CLCN1 encodes the main part of the skeletal muscle Cl- ion channel, including 23 exons, and more than 60 point mutations and 3 deletions have been found. Specifically, the protein produced by the CLCN1 gene forms a channel that controls the flow of negatively charged chlorine atoms (chloride ions) into these cells. The main function of this channel is to stabilize the cell's charge, thereby preventing abnormal muscle contraction.
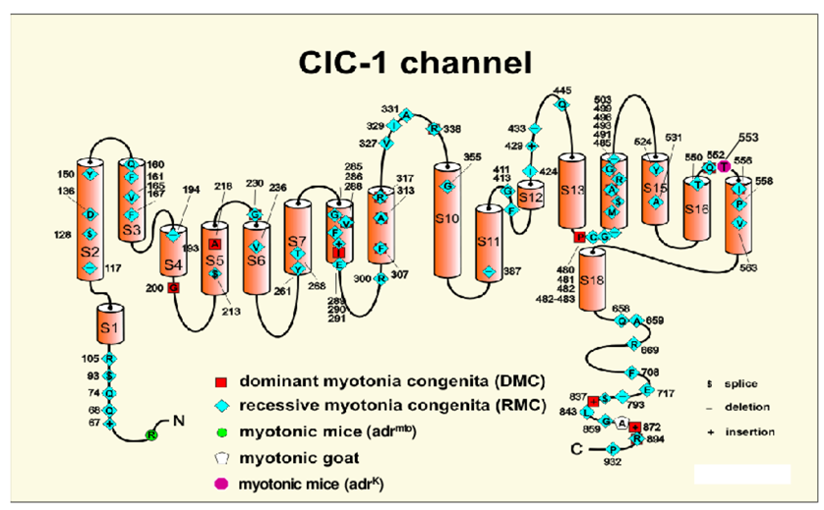
Figure 1. The model shows the skeletal muscle chloride channel monomer, ClC-1. The functional channel is a homodimer encoded by the CLCN1 gene. (Sunisa Chaiklieng, 2008)
Mutations in the CLCN1 gene can change the regular structure or function of chloride channels. The altered channel cannot properly adjust the chloride ion flow, thereby reducing the chloride ion level entering the skeletal muscle cells. The disruption of the chloride current triggers the prolonged contraction of the muscle, which is a sign of myotonic myopathies. At present, about 15% of patients with Thomsen type have Cl- ion channel gene mutations, and about 16% of Becher type patients. Because several CLCN1 mutations can cause Becker's disease or Thomson's disease, doctors usually rely on characteristic signs and symptoms to distinguish between the two forms of congenital myotonia.
Genetic Mechanism
In patients with congenital myotonia and animal models of myotonia, it was found that the Cl- ion conductance of myotonia decreased. The mutations in different forms of the amino acid gene encoding the Cl- ion channel peptide chain caused Thomsen and Becher congenital myotonia, respectively. The former is autosomal dominant inheritance, the latter is autosomal recessive inheritance, both have genetic heterogeneity. Mutations in the CLCNI gene encoding the Cl- ion channel on chromosome 7q35 cause permanent weakening of resting Cl-conduction (Cl-influx), reducing the depolarization threshold and making it tend to be myotonic myopathies.
References
- Chrestian N, et al. Myotonia congenita--a cause of muscle weakness and stiffness. Nat Clin Pract Neurol. 2006, 2(7):393-9.
- Colding-Jørgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve. 2005, 32(1):19-34.
- Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum Mutat. 2002, 19(4):423-34.
- Sunisa Chaiklieng. Low chloride conductance myotonia in vitro investigations on muscle stiffness and the warm-up phenomenon. Open Access Repositorium der Universität Ulm. Dissertation. 2008, http://dx.doi.org/10.18725/OPARU-1341.
Inquiry
