Pseudohypoaldosteronism
Pseudohypoaldosteronism is a disease similar to hypoaldosteronism. Clinically, pseudohypoaldosteronism is divided into two types, namely pseudohypoaldosteronism type 1 and pseudohypoaldosteronism type 2. Among them, pseudohypoaldosteronism type 1 (PHA1) is a disease characterized by the problem of regulating sodium content in the body. Sodium regulation is important for blood pressure and fluid balance, and it mainly occurs in the kidneys. However, sodium can also be removed from the body through other tissues, such as sweat glands and colon. Pseudohypoaldosteronism type 1 is named for its characteristic signs and symptoms similar to the symptoms of a low-level aldosterone hormone (which helps regulate sodium levels). However, people with PHA1 have higher aldosterone levels. Pseudohypoaldosteronism type 2 (PHA2) is another pseudohypoaldosteronism caused by problems with the regulation of sodium and potassium in the body. Similar to type 1, PHA2 is also involved in changes in sodium and potassium levels that are important in controlling blood pressure, and its regulation also occurs mainly in the kidneys. The difference is that PHA2 does not have excessive sodium loss.
Pseudohypoaldosteronism Type 1
- PHA1 Classification and Phenotype
There are two types of PHA1, which are distinguished clinically by their severity, the genes involved and how they are inherited. One type is called autosomal dominant PHA1 (also called renal PHA1) and is characterized by excessive loss of sodium in the kidneys. This form of disease is relatively mild and can usually improve in early childhood. The other is called autosomal recessive PHA1 (also called generalized or systemic PHA1), which is characterized by sodium loss from the kidneys and other organs (including sweat glands, salivary glands, and colon). This type of PHA1 is more serious and does not improve with age.
The earliest symptoms of both types of PHA1 are usually inability to gain weight, slow growth and dehydration. They are characterized by excessive sodium release in the urine, which leads to low blood sodium levels (hyponatremia) and high blood potassium levels (hyperkalemia). Infants with PHA1 also have high levels of acid in the blood (metabolic acidosis). Among them, hyponatremia, hyperkalemia or metabolic acidosis can cause other non-specific symptoms such as nausea, vomiting, extreme fatigue and muscle weakness in PHA1 infants. Babies with autosomal recessive PHA1 may have other signs and symptoms due to involvement of multiple organs. Due to the imbalance of salt in the body, affected individuals may experience an abnormal heartbeat (arrhythmia of the heart) or an episode of shock. They may also have repeated lung infections or skin injuries. However, adults with autosomal recessive inheritance of PHA1 usually have no other symptoms or signs.
- Pathogenesis of PHA1
The study found that mutations in one of the four genes involved in sodium regulation (NR3C2, SCNN1A, SCNN1B, or SCNN1G) lead to autosomal dominant or autosomal recessive PHA1. Among them, mutations in the NR3C2 gene lead to autosomal dominant PHA1. This gene provides guidance for the production of mineralocorticoid receptor protein. SCNN1A, SCNN1B, or SCNN1G gene mutations lead to autosomal recessive inheritance of PHA1. Each of these three genes provides instructions for preparing a part (subunit) of a protein complex called the epithelial sodium channel (ENaC).
The mineralocorticoid receptor regulates the protein in the cell membrane that controls the transport of sodium or potassium into the cell. In response to signals of low sodium levels (such as the presence of aldosterone), mineralocorticoid receptors increase the number and activity of these related proteins on certain kidney cell membranes. One of these proteins is ENaC, which can transport sodium into the cell. The other protein simultaneously transports sodium out of the cell and potassium into the cell. These proteins help keep sodium in the body through a process called reabsorption, and remove potassium from the body through a process called secretion.
Mutations in the NR3C2 gene can cause the non-functional or dysfunctional mineralocorticoid receptor protein to be unable to properly regulate the specialized proteins that transport sodium and potassium. As a result, both sodium reabsorption and potassium secretion are reduced, causing hyponatremia and hyperkalemia. Mutations in the SCNN1A, SCNN1B and SCNN1G genes result in reduced or non-functional ENaC channels. Like autosomal dominant PHA1, reduced or missing ENaC function in the kidney can lead to hyponatremia and hyperkalemia. In addition, non-functional ENaC channels in other body systems can cause other signs and symptoms of autosomal recessive PHA1, including lung infections and skin lesions.
Pseudohypoaldosteronism Type 2
- PHA2 Phenotype
People with PHA2 usually have high blood pressure, and although their kidneys are functioning normally, their blood levels of potassium are higher (hyperkalemia). The age of onset of PHA2 is variable and difficult to determine; some affected individuals are diagnosed in infancy or childhood, while others are diagnosed as adults. Hyperkalemia usually occurs first, while hypertension develops with the environment. Patients also have high levels of chloride (hyperchloremia) and acid (metabolic acidosis) in their blood (collectively referred to as hyperchlorine metabolic acidosis). People with hyperkalemia, hyperchloremia, and metabolic acidosis may experience non-specific symptoms such as nausea, vomiting, extreme fatigue (fatigue), and muscle weakness. In addition, people with PHA2 may have high levels of calcium in their urine (hypercalciuria).
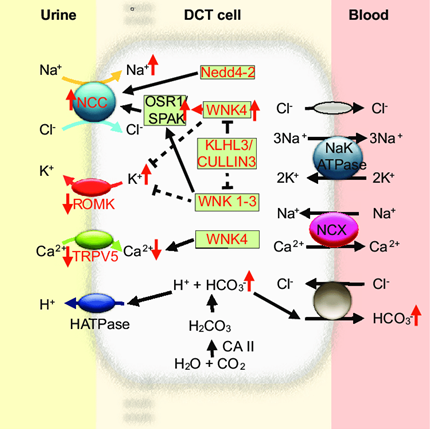
Figure 1. Molecular mechanism of pseudohypoaldosteronism type II (PHAII) in the distal convoluted tubule. (Ganesh Pathare, et al.; 2013)
- Pathogenesis of PHA2
Research results indicate that PHA2 can be triggered by mutations in the WNK1, WNK4, CUL3, or KLHL3 genes. These genes all play a role in blood pressure regulation. The proteins produced by WNK1 and WNK4 genes can control the content of sodium and potassium in the body by regulating the channels in the cell membrane. This process mainly occurs in the kidneys. Mutations in any of these genes disrupt the control of these channels, resulting in abnormal levels of sodium and potassium in the body. And then, the affected individual develops hypertension and hyperkalemia. The protein produced by the CUL3 gene (called cullin-3) and the KLHL3 gene help control the amount of WNK1 and WNK4 proteins available. Cullin-3 and KLHL3 are two fragments of the complex called E3 ubiquitin ligase. The E3 ubiquitin ligase containing cullin-3 and KLHL3 can label WNK1 and WNK4 proteins with ubiquitin, causing them to break down. Mutant genes in CUL3 or KLHL3 impair the breakdown of WNK4 protein. (The effect of these mutations on the WNK1 protein is unclear.) Excessive WNK4 may disrupt the control of sodium and potassium levels, leading to hypertension and hyperkalemia.
References
- Kolla V, Litwack G. Transcriptional regulation of the human Na/K ATPase via the human mineralocorticoid receptor. Mol Cell Biochem. 2000, 204(1-2):35-40.
- Masilamani S, et al.; Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999, 104(7):R19-23.
- Pujo L, et al.; Mineralocorticoid receptor mutations are the principal cause of renal type 1 pseudohypoaldosteronism. Hum Mutat. 2007, 28(1):33-40.
- Boyden LM, et al.; Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012 Jan 22;482(7383):98-102.
- Chávez-Canales M, et al.; WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension. 2014, 64(5):1047-53.
- Ganesh Pathare, et al.; A molecular update on pseudohypoaldosteronism type II. AJP Renal Physiology. 2013, 305(11). DOI: 10.1152/ajprenal.00440.2013.
Inquiry
