Spinocerebellar Ataxia Type-13
Spinocerebellar ataxia 13 (SCA13) is a rare subtype of spinocerebellar ataxia (SCA), a group of neurological diseases that cause degeneration of the brain and spinal cord. The age of symptoms and the type and severity of SCA13 symptoms are heterogeneous, and they may vary from person to person even in the same family. In almost every case, the disease progresses very slowly and does not affect a person's lifespan. Symptoms are most common in early childhood or late middle age, but may persist from infancy to 60 years of age. Childhood forms are usually not obvious, but they may also include mild to moderate learning problems, which take longer to learn to crawl, walk or run (slow motor skills development), or have seizures.
The main symptoms of SCA13 include difficulty coordinating movement (ataxia), especially walking; abnormal eye movements (nystagmus); mild to moderate intellectual disability; and involuntary muscle twitches (myoclonic twitches). Other symptoms may include speech difficulties (dysarthria), eating difficulties (dysphagia), slow movements (slow movement), mild bladder problems, stiffness and brisk deep tendon reflexes, and loss of vibration sensation.
SCA13 is caused by changes or mutations in the KCNC3 gene, which encodes a voltage-gated potassium channel K V3.3 and is inherited in an autosomal dominant manner. Unlike many other types of SCA, SCA13 is not caused by repeated polyglutamine amplification, but a point mutation. There are two known mutations in this gene that cause SCA13: The first mutation, R420H, is located in the S4 segment of the voltage sensing channel. Since this mutation neutralizes a site that is important for voltage sensing, it causes channel barriers. Neurons expressing such channels cannot transmit information in time with sufficient fidelity. The second mutation F448L associated with SCA13 caused changes in functional channel dynamics. Compared with the wild-type, the half activation voltage of these channels shifted by 13mV hyperpolarization. Compared with the wild type, the inactivation of these channels is also greatly slowed down. This leads to a longer time after neuron hyperpolarization, so the maximum firing rate is reduced.The former mutation that affects channel gating characteristics and neuronal excitability is related to the early-onset phenotypes, while the latter mutation that affects the current amplitude is related to the late-onset phenotypes.
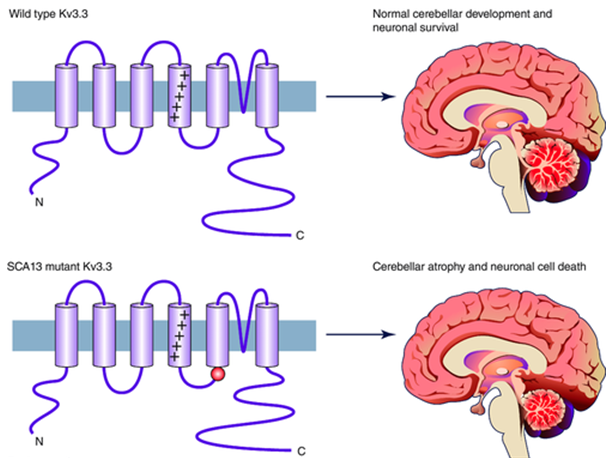
Figure 1. Kv3.3 potassium channels and spinocerebellar ataxia. ()
Like some other SCA subtypes, this gene is abundantly expressed in Purkinje cells and is believed to play an important role in promoting high-frequency action potential firing. Since the symptoms of SCA13 may be similar to other types of SCA, genetic testing can be used to confirm the diagnosis. In addition, compared with most other SCAs, brain MRI detection will find the part of the brain that helps control movement (cerebellum) loses brain cells (neurons), but the brain stem or part of the brain has no obvious brain for higher thinking (cerebral cortex) Cell loss. Treatment may include anti-epileptics, special assistance in school, speech therapy and communication equipment, and/or assistance such as crutches and walkers in the later stages of the disease. Maintaining a healthy weight can help people walk longer without help. The use of alcohol may increase the severity of uncoordinated exercise (ataxia).
References
- Ataxia espinocerebelosa tipo 13. Orphanet. 2017; http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=98768&lng=ES.
- Subramony S, et al.; Comprehensive Phenotype of the p.Arg420his Allelic Form of Spinocerebellar Ataxia Type 13. 2013, 12(3):932-936.
- Stefan-M Pulst. Spinocerebellar Ataxia Type 13. GeneReviews. 2012, http://www.ncbi.nlm.nih.gov/books/NBK1225/.
- Yalan Zhang, et al.; Kv3.3 potassium channels and spinocerebellar ataxia. J Physiol. 2016, 594.16 pp 4677-4684.
Inquiry
