Spinocerebellar Ataxia Type 6
Spinocerebellar ataxia type 6 (SCA6) is an autosomal dominant type of spinocerebellar ataxia. Studies have found that it is related to the trinucleotide CAG repeat of the gene encoding the voltage-gated calcium channel 1A subunit (CACNL1A4), showing slow progressing ataxia and dysarthria. Glutamine formed by CAG repetitive amplification caused P/Q channel dysfunction, indicating that SCA6 is a channel disease.
Clinical manifestations
The onset of SCA6 is slow, usually starting at the age of 40, and then gradually developing with age. The clinical symptoms often show cerebellar ataxia, abnormal eye movements, other accompanying signs, and repeated CAG expansion. Other manifestations related to the CACNL1A4 gene, such as migraine, periodic hemiplegia, and periodic changes in ataxia, are not seen in SCA6 patients. The first symptoms are repeated periodic transient vertigo and gait ataxia. And then, cerebellar ataxia is manifested as slow progressing gait ataxia and dysarthria, and clumsy writing. Among them, the symptom is obvious on the trunk, and the lower limbs are heavier than the upper limbs. Most patients feel stiff at the onset of the disease, but no cramps. Other accompanying symptoms are dystonic posture, involuntary movement, active knee tendon reflex, and normal plantar reflex. There may be mild peripheral neuropathy, including decreased vibration perception, decreased ankle reflex, atrophy of the abductor digitorum brevis, muscle spasm, muscle atrophy of the front legs, or paralysis during dorsiflexion of the foot. At the time of onset, upper limb ataxia and cerebellar dysarthria were mild, and gradually worsened as the course of the disease progressed. Patients with a course of more than 5 years often have symptoms of dysphagia. Most SCA6 patients have normal brainstem function 10 years before the course of the disease. Cerebellar eye movement abnormalities include gross nystagmus induced by horizontal gaze, and vertical eye movement abnormalities, such as dip nystagmus (DPN) during lateral vision, and poor vertical saccade resolution during horizontal saccade, with normal elevation or acquired increased abnormal vestibular optokinetic response (VOR) etc.. A few still have periodic alternating nystagmus (PAN), which can overlap with periodic ataxia type 2 (EA2).
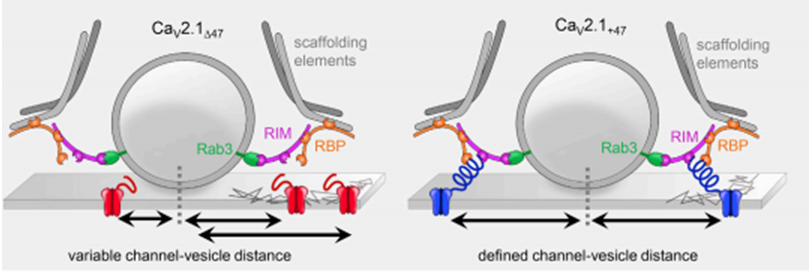
Figure 1. Schematic illustration for the organization of calcium channels in the presynaptic membrane.(Jennifer Heck, et al.; 2019)
Molecular Mechanism
Molecular biology examination shows that SCA6 has common manifestations with other polyglutamine (Gln) repeated neurodegenerative diseases encoded by CAG repeats, but the amplified fragments of SCA6 Gln are relatively small and do not form insoluble aggregates. The researchers studied the effects of 24, 30, and 40 Gln repeated amplifications in newborn hamster kidney cells on the P/Q type calcium channel current. The results found that the 24 Gln amplification channels have normal functional characteristics, while 30 or 40 Gln The amplified channels showed a voltage-dependent inactivation shift of 8 mV toward the hyperpolarization direction, and the number of channels activated by the resting membrane potential state was significantly reduced. It shows that Gln amplification can reduce the influx of calcium ions in Purkinje cells and other neurons, leading to neuronal death and cerebellar atrophy. In addition to the toxic effects of Gln amplification after aggregation, it can also directly change the function of affected gene products. Studying the expression of mutant α1A subunit in oocytes, it was found that the channel activation voltage-dependently moved 10 mV in the direction of hyperpolarization. The activation of P/Q-type channels shifts toward hyperpolarization, which can have a great impact on Purkinje cells and the ion content in cells at the same time. Such as uncompensated response, it can cause a large number of voltage-gated calcium channels to open preferentially and a large amount of calcium ion influx. Calcium ion overload can cause the activation of calcium ion-sensitive enzymes, such as caspases, calpains, phospholipases, etc., leading to cell death. Calcium ion overload can trigger neuron death, but there is evidence that lack of sufficient calcium ion influx can also cause neuron death. Studies have found that changes in the function of α1A can cause the degeneration of Purkinje cells that mainly express α1A, which is related to the voltage inactivation-dependent negative shift caused by the decrease of calcium ion influx.
References
- Ishiguro T, et al.; The carboxy-terminal fragment of alpha(1A) calcium channel preferentially aggregates in the cytoplasm of human spinocerebellar ataxia type 6 Purkinje cells. Acta Neuropathol. 2010, 119(4):447-64.
- Kordasiewicz HB, Gomez CM. Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics. 2007, 4(2):285-94.
- Rajakulendran S, et al.; Dysfunction of the Ca(V)2.1 calcium channel in cerebellar ataxias. F1000 Biol Rep. 2010, 2. pii: 4. doi: 10.3410/B2-4.
- Jennifer Heck, et al.; Transient Confinement of CaV2.1 Ca2+-Channel Splice Variants Shapes Synaptic Short-Term Plasticity. Neuron. 2019,103: 66-79.
Inquiry
