CACNA1G Voltage Sensitive Calcium Channel (VSCC) mediates the entry of calcium ions into excitatory cells, and is also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell movement, cell division and cell death. CACNA1G is located in the Moe1 locus, and RNA-seq shows strain-dependent differences in the transcriptional expression and alternative splicing of CACNA1G. CACNA1G encodes the pore-forming α1G/ Cav3.1 subunit, which is a low-pressure activated calcium channel of the T-type calcium channel family. Its subtype α-1G causes T-type calcium currents. T-type calcium channels belong to the low-pressure activation (LVA) group and are strongly blocked by mibefradil. A special feature of this type of channel is that an opening will be created at a high negative potential and will cause voltage-related inactivation. T-channels play a pacing role in central neurons and cardiac lymph node cells, and support calcium signaling in secretory cells and vascular smooth muscle. They may also be involved in the regulation of neuronal firing patterns, which are important for information processing and cell growth processes. Through clinical and animal models, it is found that there is a connection between T-shaped current and absence seizures.
CACNA1G Distribution
CACNA1G belongs to the calcium channel α-1 subunit (TC 1.A.1.11) family, and the CACNA1G subfamily. It is highly expressed in the brain, especially in the amygdala, subthalamic nucleus, cerebellum and thalamus; it is moderately expressed in the heart; it is lowly expressed in the placenta, kidneys and lungs. It is also less expressed in colon and bone marrow and tumor cells. It is highly expressed in the brain of the fetus, and is also highly expressed in the surrounding tissues of the fetus such as the heart, kidneys and lungs, suggesting that it is regulated during development. 35 human isoforms of alternative splicing have been reported.
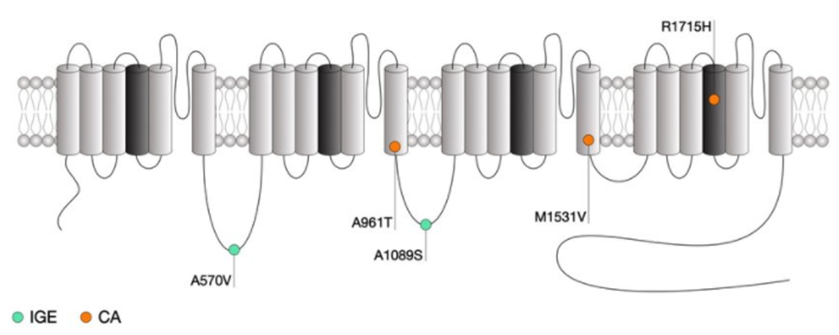
Figure 1. Location of CACNA1G mutations within the secondary structure of Cav3.1 along with their associated syndromes. (Norbert Weiss, et al.; 2019)
CACNA1G and Disease
Absence Seizure
Ca v 3.1, which is participated by CACNA1G, is a low-pressure activated calcium channel, which is also called "T-type" because of its transient opening and closing. It is expressed in the relay nucleus of the thalamic cortex and is responsible for slow wave sleep and absence seizures. In slow wave sleep, Ca v 3.1 enters a burst mode and forms a self-sustaining synchronization cycle between the cortex and the thalamus, and the sensory input is isolated from the cortex. The awake thalamus should instead transmit sensory input from outside the central nervous system. The mechanism of absence seizures has a lot in common with slow wave sleep. Therefore, the blocker Cav 3.1, which can prevent the burst mode activation of Ca, can effectively treat absence seizures. Commonly used drugs include ethosuximide and trimethyldione.
Hereditary Cerebellar Ataxia
Hereditary cerebellar ataxia (CA) is a clinically neurodegenerative disease characterized by cerebellar syndrome, usually accompanied by other neurological or non-neurological symptoms. All transmission modes have been described. In autosomal dominant CA (ADCA), more than 30 gene mutations are involved, but molecular diagnosis is still unknown in about 40% of cases. For a long time, the meaning of ion channels has been a topic in CA genetics. Recently, mutations in some channel genes have been associated with ADCA. In a large family affected by ADCA and mild pyramidal signs, researchers combined linkage analysis and whole-exome sequencing to find pathogenic variants. In CACNA1G, the researchers identified a mutation that caused the arginine in the S4 section of the voltage sensor of the T-channel protein Cav3.1 to change to histidine. Through electrophysiological experiments, the characteristics of the mutant and wild-type Cav3.1 channels were compared. It is found that the current-voltage and steady-state activation curves of the abrupt channel move in the positive direction, while the passivation curve has a higher slope. Computer modeling of deep cerebellar nucleus (DCN) neurons suggests that this mutation causes decreased neuronal excitability. The results showed that CACNA1G is highly expressed in the cerebellum, and its gene mutations can cause the up-regulation of ADCA genes.
Epilepsy
In patients with multiple epilepsy syndromes, more than 1,200 mutations in the neuronal voltage-gated sodium channel (VGSC) gene have been identified. A common feature of hereditary epilepsy is variable expression between individuals with the same mutation. The genetic mapping and RNA-Seq analysis of the SCN2A Q54 transgenic spontaneous epilepsy mouse model confirmed that Cacna1g is a candidate modifier gene of the Moe1 locus, which affects the severity of the Scn2a Q54 phenotype. In this study, the researchers tested whether transgenic alteration of Cacna1g expression would alter the severity of the Scn2aQ54 epileptic phenotype to evaluate the modification potential of Cacna1g, which encodes the Cav3.1 voltage-gated calcium channel. The results showed that the frequency of epileptic seizures in Scn2aQ54 mice increased, and the expression of Cacna1g increased; the frequency of epileptic seizures decreased, and the expression of Cacna1g decreased. These indicate that Cacna1g has the effect of regulating epilepsy regulatory genes.
References
- Kim D, et al.; Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T‐type Ca(2 + ) channels. Neuron. 2001, 31: 35– 45.
- Coutelier M, et al.; A Recurrent Mutation in CACNA1G Alters Cav3.1 T-Type Calcium-Channel Conduction and Causes Autosomal-Dominant Cerebellar Ataxia. Am J Hum Genet. 2015, 97(5):726-37.
- Kim CH. Cav3.1 T-type calcium channel modulates the epileptogenicity of hippocampal seizures in the kainic acid-induced temporal lobe epilepsy model. Brain Res. 2015, 1622: 204– 216.
- Chemin, J., et al.; Specific contribution of human T-type calcium channel isotypes (alpha-1G, alpha-1H, and alpha-1I) to neuronal excitability. J. Physiol. 2002, 540: 3-14.
- Chemin, J., et al.; De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain. 2018,141: 1998-2013.
- Coutelier, M., et al.; A recurrent mutation in CACNA1G alters Cav3.1 T-type calcium-channel conduction and causes autosomal-dominant cerebellar ataxia. Am. J. Hum. Genet. 2015, 97: 726-737.
- Norbert Weiss, et al.; Genetic T-type calcium channelopathies. Journal of Medical Genetics. 2019, 57(1):1-10.
Inquiry