CACNA1H
| Catalog | Product Name | Gene Name | Species | Morphology | Price |
|---|---|---|---|---|---|
| ACC-RI0002 | Human CACNA1H Stable Cell Line-HEK293 | CACNA1H | Human | Epithelial | INQUIRY |
Introduction of CACNA1H
CACNA1H (calcium voltage-gated channel subunit Alpha1 H) encodes the α-1 subunit family. The α-1 subunit family is a protein in the voltage-dependent calcium channel complex. Calcium ion channels mediate the influx of calcium ions into cells when the membrane is polarized, and are composed of α-1, α-2/δ, β and γ subunit complexes with a ratio of 1:1:1:1. The alpha-1 subunit has 24 transmembrane fragments and forms pores through which ions enter the cell. Each protein in the complex has multiple isoforms, which may be encoded by different genes, or may be the result of transcript splicing. Studies have shown that certain mutations in this gene can cause childhood epilepsy (CAE).
Voltage-sensitive calcium channels produce T-shaped calcium currents. T-type calcium channels belong to the "low pressure activation (LVA)" group. The special feature of this type of channel is that it has an opening at a very negative potential and has voltage-dependent inactivation. T-channels play a pacing role in central neurons and cardiac lymph node cells, and support calcium signal transduction in secretory cells and vascular smooth muscle. They may also be involved in the regulation of neuronal firing patterns. In the adrenal glomerulus, it participates in the signal transduction pathway leading to the production of aldosterone in response to AGT/angiotensin II or hyperkalemia.
CACNA1H protein is involved in T-channel formation
The T-type Ca2+ channel contains three CaV3 pore-forming channel subunits (CaV3.1, CaV3.2 and CaV3.3), which are encoded by members of the CACNA1 gene family. Three T channel genes were identified through the cloning of mammalian cDNA: CACNA1G, encoding Cav3.1; CACNA1H, encoding Cav3.2; and CACNA1I, encoding Cav3.3. The common biophysical feature of these three CaV3 channel subtypes is that they can be activated at a voltage below the threshold, showing relatively slow activation and inactivation dynamics, and complete inactivation during continuous depolarization. In addition to their common features, CaV3 channels also exhibit divergent characteristics. The inactivation kinetics of CaV3.3 channels is particularly slow, and the sensitivity of CaV3.2 channels to nickel is much higher than that of CaV3.1 and CaV3.3.
CACNA1H related diseases
Because the T-type Ca2+ channel is widely distributed and participates in a variety of in vivo mechanisms, when its constituent subunit CACNA1H is mutated, it will cause a variety of clinical diseases.
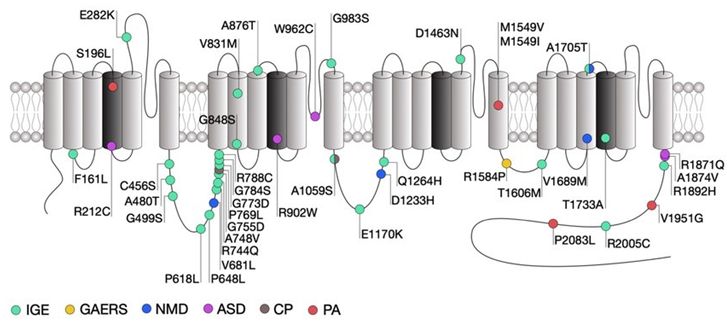
Figure 1. Location of CACNA1H mutations within the secondary structure of Cav3.2 along with their associated syndromes. (Norbert Weiss, et al.; 2019)
There are two different calcium ion channels in cardiomyocytes. L-type Ca(2+) channels are ubiquitous in cardiomyocytes and are also the main source of Ca(2+) excitation-contraction coupling and pacemaker activity. The T-type Ca(2+) channel has various functions, which are affected by mammalian species, heart area, age and various heart diseases. The two subtypes of cardiac T-type Ca(2+) channel proteins, Ca(V)3.1 and Ca(V)3.2, are functionally expressed in the embryonic heart, but are significantly reduced during development. In the adult heart, T-type Ca(2+) channels are almost undetectable in ventricular myocytes, but they are most common in the conduction system and can promote depolarization of the sinus node pacemaker. Interestingly, T-type Ca(2+) channels are re-expressed in atrial and ventricular cells under different pathological conditions such as hypertrophy and heart failure, which in turn leads to abnormal electrical activity and excitation contraction coupling. Although it is found functionally that T-type channels contribute less to the triggering of Ca(2+) release than L-type Ca(2+) channels, studies have shown that T-type Ca(2+) channels play a significant role in the process of pathological cardiac hypertrophy.
Primary aldosteronism (PA) is the most common form of secondary hypertension. Mutations in KCNJ5, ATP1A1, ATP2B3 and CACNA1D have been found in aldosterone-producing adenomas (APA) and familial hyperaldosteronism (FH). The recurrent mutation of CACNA1H (encoding Cav3.2) was identified as a family form of early-onset PA. The electrophysiological analysis of the mutant Cav3.2 channel showed that the Ca2+ current characteristics of all mutants had significant changes, indicating that the functional phenotype was acquired. Transfection of mutant Cav3.2 in H295R-S2 cells resulted in increased aldosterone production and/or expression of genes encoding steroidases after K+ stimulation.
In addition, studies have found that gain-of-function mutations in the Ca V 3.2 gene are related to idiopathic generalized epilepsy. Similarly, the increased expression of Ca V 3.2 obtained in the thalamus and hippocampus has caused chronic epilepsy.
References
- Karen M. J. van Loo, et al.; Transcriptional Regulation of T-type Calcium Channel CaV3.2. The Journal of Biological Chemistry. 2012, 287, 15489-15501.
- Kyoichi Ono, et al.; Cardiac T-type Ca2+ channels in the heart. Journal of Molecular and Cellular Cardiology. 2009, 48(1): P65-70
- E.Perez-Reyes, et al.; Molecular characterization of T-type calcium channels. Cell Calcium. 2006, 40(2): Pages 89-96.
- Norbert Weiss, et al.; Genetic T-type calcium channelopathies. Journal of Medical Genetics. 2019, 57(1):1-10.
Inquiry