Cystic fibrosis transmembrane conductance regulator (CFTR) is a unique chloride channel. Although it is called a chloride channel, it is structurally different from other chloride channels. CFTR is a member of ATP-binding cassette transport Body (ABC) family. CFTR mainly provides a selective channel for the movement of chloride ions across the epithelium, and plays an important decisive role in transepithelial salt transport, liquid flow and ion concentration adjustment. Since the CFTR gene was cloned in the late 1980s, it has been a hot spot in ion channel research.
Basic Characteristics of CFTR
In 1989, Riordan et al. first cloned the CFTR gene. It was not until 1991 that CFTR was identified as a chloride ion selective channel, and thus began a systematic study of CFTR as a chloride ion channel. The CFTR gene is located on the long arm of human chromosome 7 (7q31.2). It has a total length of 250,000 bp and contains 27 exons. The final transcribed mature mRNA has a length of 6129 bases, of which 4443 is a codable sequence. The mature CFTR protein has a full length of 1480 amino acid residues and a relative molecular mass of about 168173. In addition, CFTR has also been successfully cloned in other organisms. The mouse CFTR is located on the 6th chromosome and the rat is located on the 5th chromosome. CFTR is widely distributed. It is expressed in the cell membranes of many organs, such as the lung, liver, pancreas, intestine, and gonads. Although it is called chloride channel, it also involves the transport of other monovalent anions. Due to physiological conditions, chloride is the most important, so it is called chloride channel.
CFTR is a transmembrane protein. It is difficult to obtain ideal crystals. A complete structure image has not yet been obtained. However, because it belongs to the ABC family and the structure of some members of the ABC family has been clarified, CFTR is speculated based on the sequence alignment structure. Recently, the general crystal structure of CFTR was obtained, and its spatial structure was initially obtained using electron microscopy. It is similar in structure to another member of the ABC family of eukaryotes, P-glycoprotein, which shows the rationality of the previous speculation. It is now certain that CFTR is composed of five functional structures: two transmembrane domains (MSD1 and MSD2); two nucleotide binding domains (NBD1 and NBD2); and a regulatory domain R. Among these domains, two MSDs form selective chloride channels, an NBD domain regulates the gating of chloride channels, and phosphorylation of the R group controls channel activity.
CFTR Regulation Mechanism
The two six transmembrane domains MSD1 and MSD2 together form a channel selective to chloride ions. The narrowest part of the channel has a diameter of 0.53-0.60nm. Under normal circumstances, the channel is blocked by other large anions or regulatory domains. When the intracellular chloride ion concentration increases and cAMP-dependent protein kinase is activated, the channel can finally be opened, which effectively regulates the opening and closing of the channel. In addition, the concentration of extracellular chloride ions can also affect the gate control of the channel, and its concentration can also promote the opening of the channel. Unlike other ABC proteins, CFTR allows two-way permeation of chloride ions instead of directed transport. Part of the amino acids of the two MSDs constitute the selective transport of chloride ions, such as positively charged K95, R134, R334, K335, R347 and R1030 of CFTR are highly conserved among species, and their mutations will affect the channel to chloride ions. Because the complete structure of CFTR has not been elucidated, the selective molecular mechanism of chloride ions has not yet been fully elucidated. The gating of CFTR is mainly regulated by two NBDs, and the research on them is the most detailed. NBD contains a large number of highly conserved sequences. Each NBD domain contains a conservative phosphate binding loop (called P loop or Walker A motif), in addition to conservative Walker B motifs and LSGGQ motifs. It is speculated that these The domain plays an important role in the binding and hydrolysis of ATP.
Similar to other ATP-dependent gated channels, the binding of ATP is necessary for the opening of the CFTR channel. The binding and subsequent hydrolysis of ATP effectively regulate the gate control of the channel. Recent studies have found that ADP can inhibit the opening of the channel. NBD1 and NBD2 both contain ATP-binding domains and have ATPase activity, which can drive the opening of the channel by hydrolyzing ATP. In this process, the gating is mainly regulated by NBD adenylate kinase activity, not ATPase activity. Therefore, although a large amount of ATP is consumed, it does not consume much energy.
So how do two NBDs achieve the gate control of chloride channels under the drive of ATP? Kidd and other studies have shown that when the two domains exist alone, the ATPase activity is low, but only when the two form dimerization. The body can effectively increase enzyme activity. In particular, Vergani et al. recently discovered that when NBDI and NBD2 exist independently, the chloride channel is closed, and when a tightly bound dimer is formed, the chloride channel is opened. And ATP is required for the process of forming a dimer, so the tight dimerization of the two NBD domains driven by ATP is a prerequisite for ion channel opening, so as to achieve an organic combination of ATP hydrolysis and channel gating. So are the functions of the two domains in the formed dimer the same? The study found that both domains can bind to ATP, but only NBD2 can hydrolyze ATP to promote the opening of the channel, indicating that the two domains have their own mechanisms Completed the coupling process of ATP hydrolysis and gating.
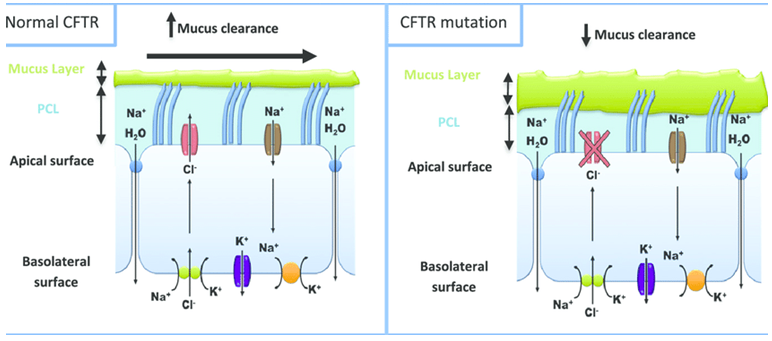
Figure 1.The defect in cystic fibrosis. (Shankar Kumar, et al.; 2014)
Compared with other members of the ABC family, CFTR is the only molecule known to contain the regulatory domain R, which can regulate the transmembrane movement of chloride ions together with the two NBD domains by adding or removing phosphoric acid. Domain R contains a large number of phosphorylated sites, such as serine and threonine, which can be phosphorylated by protein kinase A (PKA) or protein kinase C (PKC), and these sites are also regulated by phosphatase , So as to achieve reversible phosphorylation regulation. When the serine in domain R is phosphorylated, it can effectively promote the dimerization of the two NBD domains, while also increasing their ability to bind and hydrolyze ATP, thereby regulating the gate control of chloride channels.
CFTR and CF
CFTR chloride channel is the only channel named after disease among all chloride channels. It belongs to the ABC family structurally and is normally expressed on the plasma membrane of ciliated cells in a variety of glandular tissues, such as the nasal cavity, lungs, and digestive tract. In addition to its physiological function of transporting chloride ions to regulate the internal and external charge balance of cells, it also participates in the transport of other important substances, such as HCO3-, to a certain extent, so it plays an important role in the physiological functions of the body. CFTR gene mutations easily affect protein function, leading to the formation of cystic fibrosis (CFTR) (CFTR is also named for this disease). The symptoms and complications of the disease include tracheal-related diseases, pancreatic failure, and meconium Intestinal obstruction, male infertility and increased salt concentration in sweat, etc.
References
- Riordan J R, et al.; Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science.1989, 245(4922): 1066-1073.
- Vergani P, et al.; CFTR channel opening by ATP-driven tight dimerization of is nucleotide-binding domains.Nature. 2005. 433(7028): 876-880.
- Shankar Kumar, et al.; Cystic fibrosis—what are the prospects for a cure? Eur J Intern Med. European Journal of Internal Medicine. 2014, 25:803-07.
Inquiry