CLCN1
| Catalog | Product Name | Gene Name | Species | Morphology | Price |
|---|---|---|---|---|---|
| ACC-RI0082 | Human CLCN1 Stable Cell Line-CHO | CLCN1 | Human | Epithelial-like | INQUIRY |
CLCN1 gene encodes a protein involved in the formation of chloride channels. These channels transport negatively charged chloride ions and play a key role in the cell's ability to generate and transmit electrical signals. These channels only exist in the muscles used for exercise (skeletal muscles). In order for the body to function normally, skeletal muscles must be tensioned (contracted) and relaxed in a coordinated manner. The contraction and relaxation of muscles are controlled by the movement of certain ions into and out of muscle cells. The ClC-1 channel spans the cell membrane and controls the entry of chloride ions into these cells. This influx stabilizes the charge of the cells, thereby preventing abnormal muscle contraction. The ClC-1 channel is composed of two identical protein subunits, each of which is produced by the CLCN1 gene. Although each subunit forms a separate hole for chloride ions to pass through, the two proteins work together to regulate the entry of chloride ions into skeletal muscle cells.
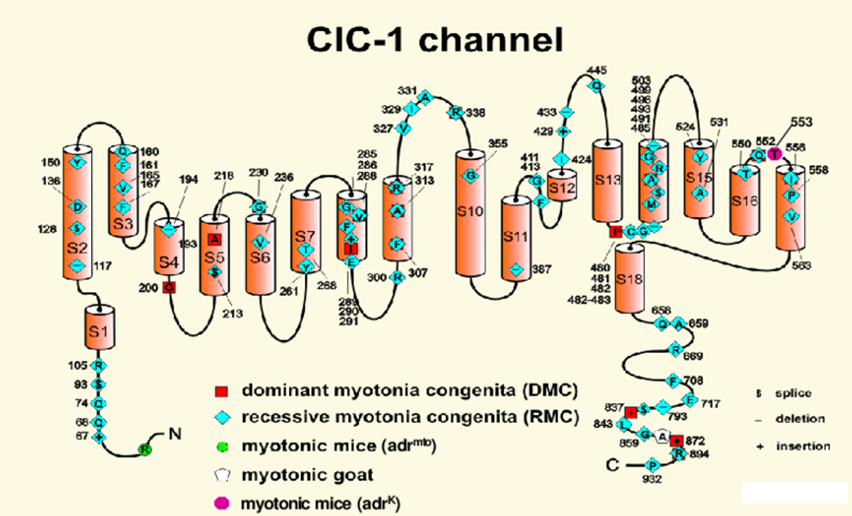
Figure 1. Membrane topology of the chloride channel.(Sunisa Chaiklieng, et al.; 2007)
The CLCN family of voltage-dependent chloride channel genes consists of 9 members (CLCN1-7, Ka and Kb), which exhibit different functional characteristics and have significant sequence homology. The protein encoded by this gene regulates the electrical excitability of the skeletal muscle membrane.
Skeletal muscle chloride channel protein is a protein encoded by the CLCN1 gene. Mutations in this protein can cause congenital muscle rigidity. CLCN1 is essential for the normal function of skeletal muscle cells. Under normal physiological conditions, for the normal activities of the body, skeletal muscles must be tense and relaxed in a coordinated manner. This change is controlled by the ions that enter and exit the muscle cells. Among them, CLCN1 is involved in the formation of such an ion channel, which controls the entry of negatively charged chloride ions into these cells. The main function of this channel is to stabilize the cell's charge and allow the muscles to contract normally. In patients with congenital myotonia caused by mutations in the CLCN1 gene, ion channels allow too little chloride ions to enter the cell. Lack of chloride ions can cause muscles to contract for a long time, which is a sign of muscle rigidity.
CLCN1 and Expression
Through homology screening with CLCN1, the main rat skeletal muscle chloride channel, Koch et al. cloned part of human CLCN1 cDNA, which covers about 80% of the coding sequence. This region is 88% identical to the rat channel in amino acid sequence. Chen et al. found that the CLCN1 gene is expressed in various human brain regions, including the cerebellum, hippocampus, spinal cord, cerebral cortex, and heart. Clcn1 is also expressed in the brain and heart of developing and adult mice. In the mouse brain, Clcn1 neuronal expression was detected in the pyramidal and dentate granular cells of the hippocampus, purkinje cells of the cerebellum, scattered brain stem nuclei, frontal neocortex and thalamus. The results indicate that CLCN1 has neuronal function and excitability in addition to its known role in skeletal muscle.
Gene Function
Steinmeyer et al. determined that CLCN1 is a homo-oligomer, most likely to have 4 subunits. Pusch et al. used Xenopus transfection to prove that four different mutations found in patients with congenital myotonia shift the gate of CLCN1 to a positive voltage. When these mutant cDNAs are co-expressed with wild-type subunits, they impose an altered voltage dependence on the heterologous channel, which then only opens within a voltage range that does not significantly promote action potential repolarization. Without this repolarization, the sodium channels have enough time to recover from inactivation, leading to the typical tonic motion, which is a series of repetitive action potentials.
CLCN1 and Disease
Myotonia Congenita
The muscle chloride channel CLCN1 regulates the electrical excitability of the skeletal muscle membrane. Skeletal muscle has an abnormally high resting Cl- conductance. In vitro studies have shown that this reduction in conductance can lead to electrical instability and muscle rigidity in human and animal models. Muscle Cl- conductance is mainly mediated by the CLCN1 chloride channel.
In patients with congenital myotonia, more than 150 CLCN1 gene mutations have been found. Most of these mutations cause an autosomal recessive inherited disease, which is called Becker's disease. Autosomal recessive inheritance means that two copies of the gene in each cell have changed. When CLCN1 mutations change the structure or function of the two protein subunits that make up the ClC-1 channel, it can cause Becker's disease. The change in the channel greatly reduces the flow of chloride ions into the skeletal muscle cells, which triggers long-term muscle contraction. Abnormal sustained muscle contractions are characteristic of muscle rigidity. CLCN1 mutations also cause congenital myotonia in an autosomal dominant form, which is called Thomson's disease. Autosomal dominant inheritance means that one copy of an altered gene in each cell is enough to cause disease. Studies have shown that the CLCN1 mutation that causes Thomson's disease changes one of the two protein subunits that make up the ClC-1 channel. The altered protein exhibits new but harmful properties, destroying the ability of the two subunits to regulate the flow of chloride ions. The reduction of chloride ions into skeletal muscle cells leads to muscle rigidity, which is the basis of stiffness and other muscle problems in patients with congenital myotonia. Because several CLCN1 mutations can cause Becker's disease or Thomson's disease, doctors usually rely on characteristic signs and symptoms to distinguish between the two congenital myotonias.
References
- Grunnet M, et al.; Characterization of two new dominant ClC-1 channel mutations associated with myotonia. Muscle Nerve. 2003, 28(6):722-32.
- Imbrici P, et al.; ClC-1 chloride channels: state-of-the-art research and future challenges. Front Cell Neurosci. 2015, 9:156.
- Jentsch TJ, et al.; Molecular structure and physiological function of chloride channels. Physiol Rev. 2002, 82(2):503-68.
- Pusch, M., et al.; Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron.1995,15: 1455-1463.
- Chen, T. T., et al.; Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology. 2013, 80: 1078-1085.
- Sunisa Chaiklieng, et al.; Low chloride conductance myotonia - in vitro investigations on muscle stiffness and the warm-up phenomenon. Khon Kaen University. 2007.
Inquiry