Gamma-aminobutyric acid receptor subunit β-3 (GABRA3) is a protein encoded by the GABRB3 gene in human cells. It is located in the 15q12 region of the human genome and spans 250kb. The gene contains 10 exons in its coding region and two additional optional first exons to encode the signal peptide. Due to the existence of alternative splicing, this gene can encode many isoform proteins, and they are all subunits of the GABA A receptor (ligand-gated ion channel). Further studies have found that β-3 subunits are expressed at different levels in different developmental stages of the cerebral cortex, hippocampus, cerebellum, thalamus, olive body and the piriform cortex of the brain. Therefore, GABRB3 deficiency is associated with many human neurodevelopmental disorders and syndromes, such as Angerman syndrome, Prader-Willi syndrome, non-syndromic maxillofacial cleft, epilepsy and autism.
GABRB3 encodes a member of the ligand-gated ion channel family. The encoded protein is one of the subunits of the multi-subunit chloride channel. Existing studies have found that the multi-subunit chloride channel is the receptor that mainly inhibits the neurotransmitter γ-aminobutyric acid in the nervous system. During development, when the GABRB3 subunit performs its normal function, its role in the GABA A receptor can ensure the proliferation, migration and differentiation of forebrain cells, so that the brain develops normally.
On the contrary, new heterozygous missense mutations in the highly conserved region of GABRB3 gene can reduce the peak current amplitude of neurons or change the dynamics of the channel. These mutations result in the loss of receptor inhibitory properties, which in turn causes neurological diseases.
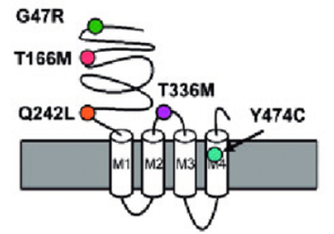
Figure 1.Schematic representation of the a 3subunit of the GABA A receptor including the predicted positions of all mutated amino acids.(C. E. Niturad et al.; 2017)
GABRA3 Related Diseases
Clinical studies have found that mutations in the GABRA3 gene may be related to the pathogenesis of Angelman syndrome, non-syndromic orofacial cleft lip, epilepsy and autism. For example, in mice, a knockout mutation of Gabrb3 causes severe neonatal death, accompanied by a cleft palate phenotype, survivors are hyperactive, lack coordination, and suffer from epilepsy. These mice also showed changes in the vestibular system in their ears, resulting in poor swimming skills, difficulty walking on the grid floor, and were found to run in irregular circles.
- Angerman Syndrome
Angelman syndrome is a genetic disease. It can cause developmental delays, speech and balance problems, mental retardation, and sometimes seizures. Studies have found that the deletion of the GABRB3 gene can cause human Angelman syndrome, and the key factors of the disease depend on the source of the missing parent. Deletion of the paternal allele of GABRB3 has no known effect on the syndrome, while deletion of the maternal GABRB3 allele leads to the development of the syndrome.
- Non-syndromic Orofacial Fissure
There is a strong link between GABRB3 expression level and proper upper jaw development. Therefore, the abnormal expression of GABRB3 may be related to non-syndromic cleft lip deformity (with or without cleft palate). Cleft lip and palate are also observed in children containing duplication of the GABRB3 locus. Knockout of the beta-3 subunit in mice can cause secondary cleft palate. The Gabrb3 transgene can be inserted into the mouse genome to restore normal facial features, indicating that the Gabrb3 gene is mainly responsible for the formation of cleft palate.
- Autism Spectrum Disorders
In some patients diagnosed with autism, there is a duplication of the Prader-Willi/Angelman syndrome region (also called the imprinted region) (15q11-13), which contains the GABRB3 gene. These patients show classic symptoms associated with the disease. The 15q11-13 regional duplication shown in patients with autism almost always originates from the mother (not the father) and accounts for 1-2% of diagnosed cases of autism. This gene is also a candidate for drug targets for autism, because when used to treat epilepsy and anxiety disorders, benzodiazepines have a physiological response to GABA-A receptors.
- Epilepsy/absence Epilepsy in Children
In animal models and human syndromes, epilepsy is often associated with defects in GABA transmission. Patients who were diagnosed with Angelman syndrome and deleted the GABRB3 gene exhibited seizures. Decreased expression of β-3 subunit may be a potential cause of absence epilepsy in children.
References
- Cook EH, et al.; Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. American Journal of Human Genetics. 1998, 62 (5): 1077-1083.
- DeLorey TM, et al.; Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behavioural Brain Research. 2008,187 (2): 207-220.
- Scapoli L, et al.; Linkage disequilibrium between GABRB3 gene and nonsyndromic familial cleft lip with or without cleft palate. Human Genetics. 2002, 110 (1): 15-20.
- Buxbaum JD, et al.; Association between a GABRB3 polymorphism and autism. Molecular Psychiatry. 2002, 7 (3): 311-316.
- DeLorey TM, et al.; GABA and epileptogenesis: comparing gabrb3 gene-deficient mice with Angelman syndrome in man. Epilepsy Research. 1999, 36 (2-3): 123-132.
- C. E. Niturad et al.; Rare GABRA3 variants are associated with epileptic seizures, encephalopathy and dysmorphic features. BRAIN. 2017, 140(11): 1-16.
Inquiry