KCNA2 gene belongs to the KV1 family of voltage-gated potassium channels, which will be expressed in the central nervous system. Kv1.2 channels belong to the delayed rectification category of potassium channels, which can achieve effective neuronal repolarization after an action potential.
KCNA2 gene location
In 1990, the researchers mapped the human homologue of the mouse MK2 K+ voltage-gated channel gene to chromosome 12 through the study of somatic cell hybridization. However, it was later discovered that this localization gene was Kcna6. After further research, it was found that Kcna2 was localized on human chromosome 1, and its mouse homologous region was mapped to mouse chromosome 3. Finally, based on the alignment of the KCNA2 sequence with the genome sequence, the scientists mapped the KCNA2 gene to chromosome 1p13.3.
KCNA2 Gene Function
Voltage-gated potassium channels mediate transmembrane potassium transport in excitable membranes, mainly in the brain and central nervous system, but also in the cardiovascular system. In normal physiological processes, this channel is mainly involved in preventing abnormal action potential discharges and in regulating neuronal output. The potassium ion selective channel formed by the KCNA2 gene is a tetramer, which can be regulated according to the electrochemical gradient of potassium ions on both sides of the membrane. The channel alternates between open and closed conformations in response to the voltage difference across the membrane. Functional homotetramer channels and heterotetramer channels can be formed, which contain different ratios of KCNA1, KCNA2, KCNA4, KCNA5, KCNA6, KCNA7, and other possible family members. Among them, channel properties depend on the type of alpha subunit that is part of the channel, and channel properties are regulated by cytoplasmic β subunits, which regulate the subcellular location of α subunits and promote the rapid inactivation of delayed rectifier potassium channels. In the body, the membrane may contain a mixture of heteromeric potassium channel complexes, so it is difficult to assign the current observed in intact tissues to any specific potassium channel family member. The homotetramer KCNA2 forms a delayed rectifier potassium channel, which opens in response to membrane depolarization and then slowly and spontaneously closes the channel. In contrast, the heteromultimer formed by KCNA2 and KCNA4 showed rapid inactivation. It can regulate neuronal excitability and act as a pacemaker in regulating neuronal action potentials. Channels containing KCNA2 play a presynaptic role and prevent excessive excitement and abnormal action potential discharge. The response to selective toxins containing KCNA2 potassium channels indicated that in Purkinje cells, the subthreshold KCNA2 potassium channels of dendrites block random spontaneous calcium peaks, inhibit dendritic overexcitation without hindering somatic action potentials. Produced to complete coordinated movement. In addition, it can also play a role in the induction of long-term enhancement of the excitability of hippocampal CA3 neurons, and can act as a downstream effector of G protein-coupled receptors and inhibit GABA input to basolateral amygdala neurons.
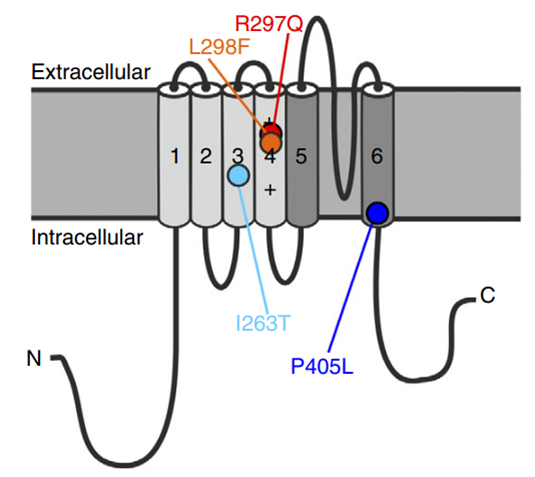
Figure 1. Structure of the voltage-gated potassium channel KV1.2. (Steffen Syrbe, et al.; 2015)
Further studies have found that the potassium channel that the protein participates in may also help regulate the neurotransmitter and the axon release of the neurotransmitter dopamine. Reduced KCNA2 expression plays a role in the perception of neuropathic pain after peripheral nerve injury, but not acute pain. Play a role in regulating non-rapid eye movement (NREM) sleep time.
KCNA2 and Disease
Achieve effective neuronal repolarization after the action potential. Loss-of-function mutations can predict over-excited neuronal membranes and repetitive neuronal firing caused by impaired repolarization. This hypothesis was confirmed by the epileptic phenotype of Kcna2 knockout mice. Mild to severe epileptic encephalopathy has been found in cases of KCNA2 de novo mutation. Phenotypes associated with its dominant negative loss of function mutations also include infant or early childhood epileptic seizures and frequent febrile and afebrile focal motor and cognitive impairment episodes that overlap with Dravet syndrome and MAE.
References
- Grabe, M., et al.; Structure prediction for the down state of a potassium channel voltage sensor. Nature. 2007, 445: 550-553.
- Grissmer, S., et al.; Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc. Nat. Acad. Sci. 1990, 87: 9411-9415.
- Gu, C., et al.; A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science. 2003, 301: 646-649.
- Hattan, D., et al.; Tyrosine phosphorylation of Kv1.2 modulates its interaction with the actin-binding protein cortactin. J. Biol. Chem. 2002, 277: 38596-38606.
- Klocke, R., et al.; Chromosomal mapping in the mouse of eight K(+)-channel genes representing the four Shaker-like subfamilies Shaker, Shab, Shaw, and Shal. Genomics. 1993, 18: 568-574.
- Steffen Syrbe, et al.; De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nature Genetics. 2015, 47: 393-399.
Inquiry