The CLCN2 gene belongs to the CLC family of genes and guides the coding of proteins involved in the formation of chloride channels. These channels transport negatively charged chlorine atoms (chloride ions) and play an important role in the cell's ability to generate and transmit electrical signals. Among them, some chloride ion channels are involved in regulating the flow of chloride ions across the cell membrane, while others transport chloride ions within the cell. The chloride channel that the CLCN2 gene participates in is called ClC-2. These channels are embedded in the outer membrane of most cells, and they transport chloride ions in and out of the cell. The function of this channel is considered to be particularly important in the nerve cells (neurons) of the brain. The ClC-2 channel regulates the size (volume) of neurons through the uptake and release of water and the maintenance of the normal balance of ions in the cell.
CLCN2 Cloning and Expression Distribution
Cid et al. cloned the human homologue of the rat voltage-gated chloride channel CLC2 from the T84 epithelial cell cDNA library. The predicted 898 amino acid protein has more than 93% homology with the rat sequence. Scholl et al. found that the CLCN2 gene is mainly expressed in the glomerular band of the human adrenal cortex. In mice, Fernandes-Rosa et al. found CLCN2 gene expression in the adrenal gland and brain. Based on the alignment of the CLCN2 sequence with the genome sequence (GRCh38), Stumpf locates the CLCN2 gene on chromosome 3q27.1.
CLCN2 Gene Function
Performing whole-cell patch-clamp analysis on cells overexpressing CLC2, the researchers found that hyperpolarization-activated chloride current (HACC) showed a time- and voltage-dependent activation and inward rectification steady-state current-voltage relationship. Decreasing the extracellular pH to 5.0 resulted in a significant increase in HACC in overexpressing cells and resulted in strong currents in parental cells from cystic fibrosis patients. Antisense CLC2 cDNA stably transfected cells, the expression of CLC2 in the parent cell was reduced, and the number of pH-dependent HACC was significantly reduced.
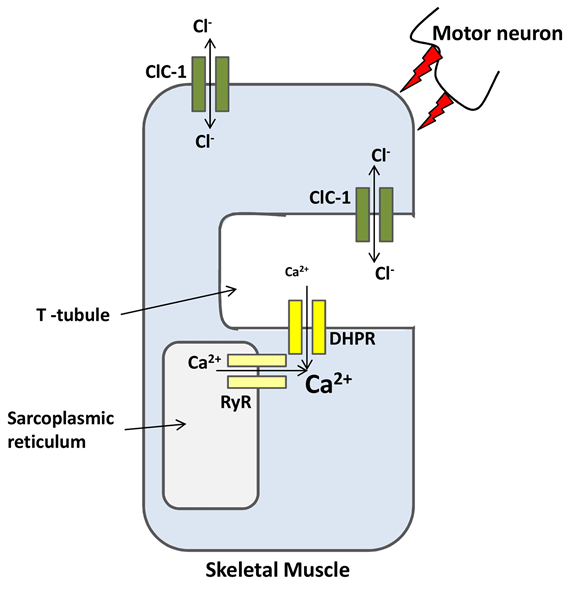
Figure 1. ClC-1 is a major ion channel involved in the membrane resting potential of skeletal muscles. (Diogo R. Poroca, et al.; 2017)
The chloride homeostasis of neurons and non-neuronal cells is partly maintained by the chloride conductance of CLCN2 channels. By immunostaining, it was found that CLCN2 channels were located in the plasma membranes of hippocampal pyramidal cells and non-pyramidal cells, dendrites, axons and somatic cells. In addition, the tails and feet of astrocytes in nerve fibers and around small blood vessels have strong immunoreactivity. Studies have found that CLCN2 participates in the transmembrane chloride movement related to GABA synaptic transmission. Further research found that the CLCN2 channel acts as a chloride ion efflux pathway, establishing and maintaining the high transmembrane chloride gradient necessary for the inhibitory GABA response. In addition, scientists also found that the CLCN2 gene is expressed on the surface of the cell body and is present in almost all GFAP-positive fibrous astrocytes in the hind limbs of the internal capsule. CLCN2 expression can also be seen in axons, oligodendrocytes and septal membrane. CLCN2 was not detected in the nucleus surrounding the neuron. Electron microscopy confirmed that CLCN2 exists in white matter astrocytes and is enriched in the process of astrocyte-astrocyte and astrocyte-axon myelin contact cells. Immune reactivity is also seen in the end products of astrocytes surrounding blood vessels.
CLCN2 and Disease
CLCN2-Related Leukoencephalopathy
At least 18 CLCN2 gene mutations have been found to cause CLCN2-related white matter lesions. The main feature of this condition is coordination and balance problems (ataxia), but it can also lead to learning disabilities, frequent headaches and vision problems. Some CLCN2 gene mutations change the single protein building block (amino acid) of the ClC-2 channel, destroy the stability of the channel, and reduce the channel function. Other CLCN2 gene mutations lead to complete loss of channel function, usually resulting in the production of abnormal short channel proteins. The shortened protein is either trapped inside the cell and cannot reach the cell membrane, or it is quickly broken down. Due to the decrease in ClC-2 channel activity, certain brain cells and the myelin surrounding neurons are filled with too much water and cannot work properly. Fluid-filled myelin cannot effectively transmit nerve impulses, leading to nervous system problems such as ataxia and other signs and symptoms of clcn2-related leukoencephalopathy.
Familial Hyperaldosteronism Type 2
The researchers identified the CLCN2 gene (5 different heterozygous missense mutations) in 8 unrelated family members with type 2 familial hyperaldosteronism. The mutation in the first family was discovered by exome sequencing and confirmed by Sanger sequencing. By screening the CLCN2 gene in 80 patients with similar phenotypes, subsequent CLCN2 mutations in other families were found. In 2 patients, CLCN2 mutations occurred de novo. This mutation was found in 4 unrelated families, and haplotype analysis showed that the mutation occurred independently. In vitro functional expression studies in human HEK293 and H295R human adrenocortical cancer cells showed that all mutants shifted the activation curve of the channel to more positive voltages and had a higher probability of opening at the glomerular resting potential . Except for one variant, all other variants modified the public door by increasing the minimum opening probability and accelerating activation. Compared with the wild type, the chloride discharge increased significantly. The S865R variant may have a regulatory function and have a similar overall effect of increasing chloride flux. The mutation increased the expression of CYP11B2 and its upstream regulator NR4A2, thereby increasing the production of aldosterone. Current clamp records show that R172Q significantly amplifies the depolarization of H295R-derived cells compared to wild-type. These findings prove the role of anion channels in the measurement of glomerular membrane potential and the production of aldosterone, and further show that CLCN2 mutations can increase excitatory anion efflux by modifying the voltage-dependent opening of the channel, thereby gaining functional enhancement.
References
- Cid, L. P., et al.; Cloning of a putative human voltage-gated chloride channel (CLC-2) cDNA widely expressed in human tissues. Hum. Molec. Genet.1995, 4: 407-413.
- Depienne, C., et al.; Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 201312: 659-668.
- Fernandes-Rosa, F. L., et al.; A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nature Genet. 2018, 50: 355-361.
- Gaitán-Peñas H, et al.; Leukoencephalopathy-causing CLCN2 mutations are associated with impaired Cl(-) channel function and trafficking. J Physiol. 2017 Nov 15;595(22):6993-7008.
- Stölting G, et al.; CLC channel function and dysfunction in health and disease. Front Physiol. 2014, 5:378.
- Wang H, et al.; Research and progress on ClC-2 (Review). Mol Med Rep. 2017, 16(1):11-22.
- Diogo R. Poroca, et al.; ClC Channels and Transporters: Structure, Physiological Functions, and Implications in Human Chloride Channelopathies. Front Pharmacol. 2017, 8: 151.
Inquiry