Potassium channels are a very diverse group of ion channels, and the subunits encoded in the channel-related gene families can be combined. This diversity contributes to the ability of specific neurons to respond uniquely to different inputs. One of these genomes, the Kv family, encodes subunits of tetrameric voltage-gated K+ channels, and is divided into several subfamilies based on sequence similarity and evolutionary relationship. Subunits of the same subfamily can form heteromultimeric channels, suggesting that each set of genes encodes a distinct set of channels. One of the Kv subfamily known as Kv3 is of particular interest because their members exhibit large unit conductances, high activation thresholds, and very fast activation and deactivation kinetics. Kv3-type channels are involved in the rapid repolarization of action potentials (APs), and their presence in neurons correlates with narrow APs, rapid post-hyperpolarization (AHP), and high-frequency firing beyond 200 Hz.
The Kv3 subfamily consists of four genes (Kv3.1 (also known as KCNC1), Kv3.2, Kv3.3, and Kv3.4). Each Kv3 gene encodes multiple products through alternative splicing at the 3' end, resulting in the expression of multiple different K+ channel proteins in the shortened C-terminal sequence alone. Although Kv3.1 and Kv3.3 subunits are expressed throughout the brain, their expression is restricted to distinct neuronal subsets. In the neocortex, thalamus, hippocampus, and striatum, Kv3-type channels are present in inhibitory GABAergic interneurons that also express the calcium-binding protein parvalbumin, a marker for rapidly spiking neurons.
Studies have shown that KV 3.1 belongs to the delayed rectifier class of channel proteins and is an integral membrane protein that mediates voltage-dependent potassium permeability of excitable membranes. The channel opens in response to stimulation by a voltage difference across the membrane, thereby forming a potassium-selective channel through which potassium ions can pass according to their electrochemical gradient. KV 3.1 channels are prominently expressed in high-frequency firing CNS neurons and are important for auditory and rapidly stimulating GABAergic interneurons, retinal ganglion cells. In addition, it regulates the action potential duration in presynaptic terminals. K V 3.1 can form functional homotetrameric channels and heterotetrameric channels containing variable ratios of ion channels.
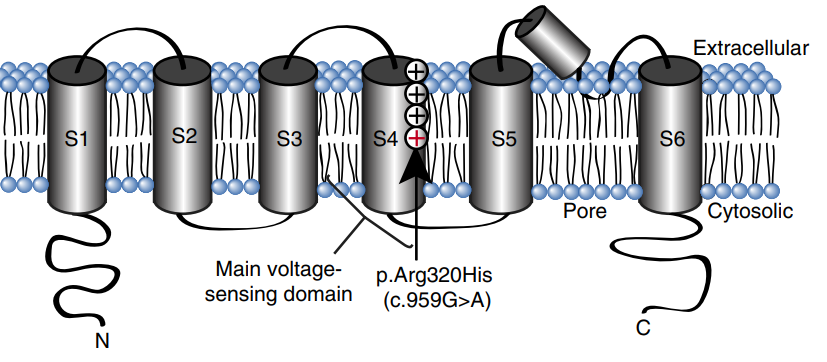
Figure 1. A schematic showing the domain structure of a single KV3.1 subunit. (Muona, Mikko; et al.; 2014)
The KCNC1 channel is a brain-expressed voltage gated potassium channel working as a “delayed rectifier”.
KCNC1 and disease
KCNC1 encodes the voltage-gated potassium channel KV 3.1, which is important for activating high-frequency firing of interneurons. Mutations in KCNC1 can lead to severe neurological deficits, including intellectual disability, epilepsy, and ataxia. Variation in KCNC1 Arg320His, which occurs in the voltage-sensing domain of the channel, causes a highly osmotic and specific form of progressive myoclonic epilepsy with severe ataxia called myoclonic epilepsy and ataxia. By expressing mutants of KV 3.1 in a knockout model that greatly reduced the excitability of mature cortical interneurons, cells expressing these channels also appeared unable to support high-frequency firing. In additon, the mutant channel also had unexpected effects on morphology, severely impairing neurite development and interneuron viability, an effect that could not be restored by blocking KV channels.
Progressive myoclonic epilepsy (PME) is a group of seizure disorders characterized by progressive neurological deficits, ataxia, myoclonus, and recurrent seizures. There are several forms of PME, of which MEAK (potassium channel mutation)-myoclonic epilepsy and potassium channel mutation-induced ataxia have recently been studied. This particular subtype is caused by recurrent de novo heterozygous mutations in the KCNC1 gene. Loss of Kv3 function disrupts the firing properties of fast-spiking neurons, affects neurotransmitter release and induces cell death. Specific to Kv3.1 dysfunction, the most affected neurons include inhibitory GABAergic interneurons and cerebellar neurons. Damage to the former cells is thought to cause myoclonus and seizures, while dysfunction of the latter causes ataxia and tremors. Phenotypically, MEAK patients generally have normal early development. Myoclonus manifests between 6 and 14 years of age, and gradually worsens over time. Tonic-clonic epilepsy may or may not be present, and some patients develop mild cognitive impairment after a seizure. Typical EEG features include generalized epileptiform discharges and, in some cases, photosensitivity. Brain imaging is normal or shows cerebellar atrophy.
References
- Poirier K, et al.; Loss of Function of KCNC1 is associated with intellectual disability without seizures. Eur J Hum Genet. 2017, 25(5):560-564.
- Carpenter, JC, et al.; Progressive myoclonus epilepsy KCNC1 variant causes a developmental dendritopathy. Epilepsia. 2021; 00:1–12.
- Muona M, et al.; A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015, 47(1):39-46.
- Muona, Mikko; et al.; A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nature Genetics. 2014, 47(1), 39–46.
Inquiry