KCNC3
| Catalog | Product Name | Gene Name | Species | Morphology | Price |
|---|---|---|---|---|---|
| ACC-RI0039 | Human KCNC3 Stable Cell Line-CHO | KCNC3 | Human | Epithelial-like | INQUIRY |
Potassium voltage-gated channel member 3, also known as Kv3.3, is a protein encoded by KCNC3 in humans.
The Kv3.3 channel subunit is one of the four members of the Kv3 voltage-dependent potassium channel family. Similar to the other members of the family Kv3.1, Kv3.2 and Kv3.4, Kv3.3 channels are also activated at positive membrane potentials. Existing studies have shown that their main function is to drive action potentials during the repolarization phase. It is well known that mutations in human ion channel genes can cause excitability disorders and dyskinesias, but it is not common for ion channel diseases to cause neurodegeneration. Among them, the more obvious exception is spinocerebellar ataxia type 13 (SCA13) caused by mutations in Kv3.3.
Like other Kv3 family channels, Kv3.3 voltage-dependent K+ channels belong to a class of ion channels called "high threshold" or "high voltage activation". These are classic delayed rectifier channels that only start to activate at potentials above -20 mV. Therefore, their contribution to resting potassium conductance is small, and they are only activated during the rising phase of the action potential. In addition, the Kv3.3 channel can also respond to rapid voltage changes. Therefore, these channels produce very rapid action potential repolarization with little or no relative refractory period, allowing neurons expressing these channels to emit a series of action potentials at a high frequency. In response to continued depolarization, the Kv3.3 channel experienced slow and partial inactivation, with a time course of hundreds of milliseconds. This inactivation occurs through the N-type inactivation mechanism. Deleting the cytoplasmic N-terminus of the channel eliminates the inactivation. The inactivation rate can also be adjusted by activating protein kinase C. There is evidence that phosphorylation of the two serine residues at the N-terminus of Kv3.3 prevents the inactivation of the N-terminus domain. As expected by its electrophysiological properties, Kv3.3 channels are expressed in neurons that fire at a high rate, especially in the brainstem and cerebellum. High levels were also found in cortical interneurons containing inhibitory paralbumin and GABAergic interneurons in other parts of the nervous system. Many of these neurons also express Kv3.1 channel, and their high-pressure activated channels may be heteromeric channels containing Kv3.3 and Kv3.1. Kv3.3 is also strongly expressed in most auditory brainstem nuclei, where neurons may emit at frequencies as high as 600-800Hz to process auditory information. Due to their high levels in Purkinje cells of the cerebellum, a large number of studies have focused on the characteristic and complex peaks produced by the Kv3.3 channel. These peaks are evoked and stimulated by these cells to cause synapses to attach to the tree through a single action potential. In addition, the Kv3.3 channel is also necessary to produce rapidly repeating spikelets, which usually occur in the process of complex spikes.
KCNC3 and Disease
Spinocerebellar Ataxia
Spinocerebellar ataxia (SCA) is one of a group of hereditary diseases characterized by slow progressive gait incoordination, usually related to poor coordination of hand, language, and eye movements. Studies have found that mutations in the Kv3.3 channel can cause spinocerebellar ataxia. Mutations in the human Kv3.3 gene (KCNC3) have been identified as the cause of spinocerebellar ataxia (SCA13). Existing studies have described two missense mutations, both of which may disrupt the role of Kv3.3 channels in Purkinje cells (and other neurons) action potential repolarization and change their output characteristics.
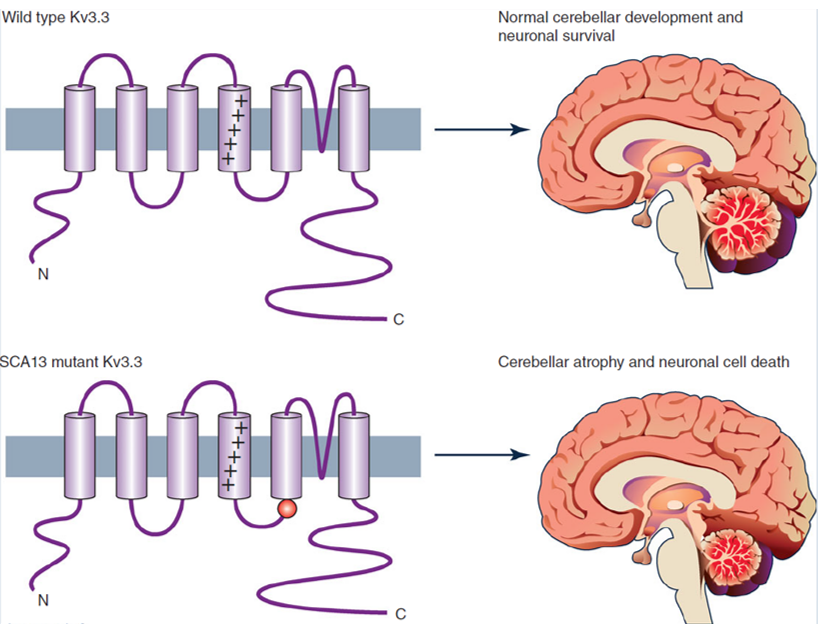
Figure 1. Kv3.3 potassium channels and spinocerebellar ataxia. (Zhang Y, et al.; 2016)
Functional research was carried out by breeding knockout mice of three Kv3 genes (Kv3.1–Kv3.3) prominently expressed in CNS neurons. These results showed that Kv3.1 and Kv3.3 knockout mice showed changes in motor behavior. For example, Kv3.1 knockout mice had increased motor activity and lack of sleep; Kv3.3 knockout mice showed impaired gait and decreased athletic ability. This is related to the abnormal discharge of Purkinje cells and the abnormal characteristics of the olive cerebellar system; and, in the case of Kv3.1 and Kv3.3 double mutants, severe ataxia, tremor and myoclonus will be exhibited.Kv3.2 knockout mice have altered EEG rhythms, increased cortical excitability, and increased susceptibility to epilepsy. This may be due to the changes in cortical structure that GABA can control.
References
- Zhang Y, Kaczmarek LK. Kv3.3 potassium channels and spinocerebellar ataxia. J Physiol. 2016, 594(16):4677-84.
- Rudy B & McBain C. Kv3 channels: Voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001, 24, 517–526.
- Desai R, et al.; Protein kinase C modulates inactivation of Kv3.3 channels. J Biol Chem. 2008, 283: 22283–22294.
- Puente N, et al.; Precise localization of the voltage-gated potassium channel subunits Kv3.1b and Kv3.3 revealed in the molecular layer of the rat cerebellar cortex by a pre-embedding immunogold method. Histochem Cell Biol. 2010, 134, 403–409.
- Nowak A, et al.; Kv3.1b and Kv3.3 channel subunit expression in murine spinal dorsal horn GABAergic interneurones. J Chem Neuroanat. 2011, 42, 30–38.
Inquiry