KCND3
| Catalog | Product Name | Gene Name | Species | Morphology | Price |
|---|---|---|---|---|---|
| ACC-RI0140 | Human KCND3 Stable Cell Line-CHO | KCND3 | Human | Epithelial-like | INQUIRY |
| ACC-RI0141 | Human KCND3 Stable Cell Line-HEK293 | KCND3 | Human | Epithelial | INQUIRY |
KCND3 gene encodes Kv4.3, which is an α subunit of the Shal family of a-type voltage-gated potassium channels, which is very important in the membrane repolarization of excitatory cells.
KCND3 Cloning and Expression
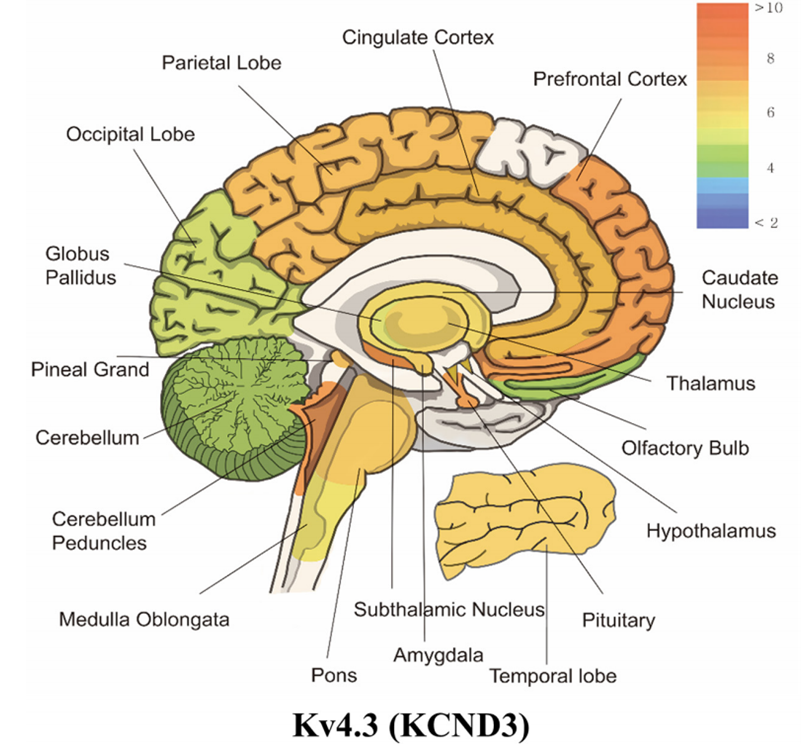
Figure 1. mRNA expression levels of panel kcnc3 (Kv4.3). (Wonjun Noh, et al.; 2019)
By screening the heart cDNA library and RT-PCR of human ventricular RNA, the researchers isolated the cDNA encoding KCND3. Deduced a protein containing 637 amino acids, this protein has 99% sequence homology with the rat homologue. Structural analysis shows that KCND3 contains 6 transmembrane fragments and intracellular N-terminal and C-terminal. RT-PCR and sequence analysis confirmed the existence of the splice variant KCND3L, whose insert encodes an additional 19 amino acids and contains a phosphorylation site. Among them, the shorter isoform was named KCND3S. Further analysis found that the KCND3L, KCND1 and KCND2 proteins have 71% homology, with the C-terminus being the least conservative. Through Northern blot analysis, the scientists detected the expression of the 8.5-kb transcript, which has the highest content in the brain and heart, but not in the kidney, liver, lung, pancreas, spleen or skeletal muscle. Further analysis in the brain found that the protein is most expressed in the cerebral cortex. Through RT-PCR analysis, they found that long transcripts are dominant in the thalamus, caudate nucleus, white matter, and epiphyses, while short transcripts are more abundant in the frontal cortex, occipital lobe, and cerebellar cortex. In the heart, only the long form of KCND3 is expressed in the heart. Through the analysis of the gene sequence, it is found that the long form of the KCND3 gene contains 7 exons with a span of at least 25 kb. The shorter isoform is encoded by 6 exons. These splice variants have no difference in voltage dependence or inactive dynamics in the basal state.
KCND3 Gene Function
The instantaneous outward potassium current I(to) is especially important in the early repolarization of many species, including humans, because it sets the platform voltage for the action potentials of the atria and ventricles. KCND2 and/or KCND3 responsible for the main alpha subunit of I(to). In the human ventricle, KCND3 is the gene encoding the K+ channel that is the basis of I(to).
KCND3 and Disease
Spinocerebellar Ataxia 19
Spinocerebellar ataxia is the main type of hereditary ataxia. The common features are the onset of middle-aged age, autosomal dominant inheritance and ataxia. In addition to cerebellar ataxia, the clinical manifestations may be accompanied by eye movement disorders, slow eye movement, optic atrophy, retinitis pigmentosa, pyramidal tract signs, extrapyramidal signs, muscle atrophy, peripheral neuropathy, and dementia.
In 19 patients with spinocerebellar ataxia, the researchers found a heterozygous 3-bp deletion in the KCND3 gene. The mutations discovered by exome sequencing and confirmed by Sanger sequencing have been isolated in the affected family. In HEK293 cells, the mutant protein showed no discernible cell surface expression and appeared to be abnormally retained in the endoplasmic reticulum. Voltage clamp recordings showed that the outward potassium current in response to voltage was reduced compared to wild-type cells. In addition, three other heterozygous missense variants (G345V, V338E or T377M) were found in the KCND3 gene in other patient families.
Brugada Syndrome 9
Brugada syndrome is a syndrome caused by abnormal function of ion channels caused by mutations in genes encoding myocardial ion channels. Clinically, this syndrome is characterized by ST-segment elevation in leads V1-3, ST-segment changes in leads V1-3, no obvious abnormalities in cardiac structure, polymorphic ventricular tachycardia (ventricular tachycardia) or ventricular fibrillation (ventricular fibrillation) ) And recurrent episodes of syncope, and sudden cardiac death.
The researchers analyzed the candidate gene KCND3 in 86 patients with Brugada syndrome who were known to have negative mutations in Brugada-related genes, and found heterozygous missense mutations in two unrelated patients, L450F and G600R. Functional analysis showed that both variants are gain-of-function mutations.
References
- Chung, M., et al.; A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain. 2003,126: 1293-1299.
- Dilks, D., et al.; Cloning and expression of the human Kv4.3 potassium channel. J. Neurophysiol. 1999, 81: 1974-1977.
- Dixon, J. E., et al.; Role of the Kv4.3 K+ channel in ventricular muscle: a molecular correlate for the transient outward current. Circ. Res. 1996, 79: 659-668.
- Duarri, A., et al.; Mutations in potassium channel KCND3 cause spinocerebellar ataxia type 19. Ann. Neurol. 2012, 72: 870-880.
- Giudicessi, J. R., et al.; Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Hum. Mutat. 2012, 33: 989-997.
- Giudicessi, J. R., et al.; Transient outward current (I-to) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011, 8: 1024-1032.
- Wonjun Noh, et al.; Transient Potassium Channels: Therapeutic Targets for Brain Disorders. Frontiers in Cellular Neuroscience. 2019, 13: 265.
Inquiry