Voltage-gated potassium channels are involved in the regulation of multiple neuronal properties, such as interspike membrane potential, action potential waveform, and firing frequency, and play an important role in controlling cell excitability in the nervous system. These channels form an evolutionarily related superfamily that can be divided into two groups, the Shaker family and the ether-a-go-go (eag) family. The eag family includes Drosophila eag, human eag, rat Eag, Drosophila eagle-related gene (erg), human ERG (KCNH2), human BEC1 (KCNH3) and human BEC2. Members of the eag family are associated with cyclic nucleotide-gated cation channels, hyperpolarization-activated cation channels, and plant hyperpolarization-activated potassium channels. A common feature at the C-terminus of Eag-type channels is a putative cyclic nucleotide binding (CNB) domain.
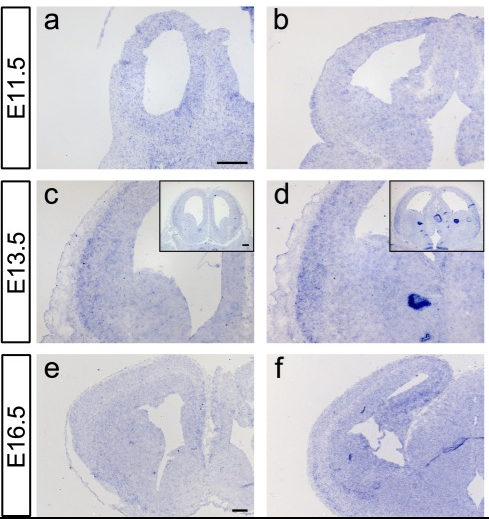
Figure1. Kcnh3-expressing regions were identified by in situ hybridization on sections from E11.5, E13.5and E16.5 murine forebrains. (Vezzali R, et al.; 2016)
KCNH3, also known as ether-a-go-go-like 2 (Elk2) or Kv12.2. Unlike Kv5. x, Kv6. x, Kv8. x, and Kv9. x, Kv12.2 can act as a modifier of other Kv channels and can generate functional channels by itself when Kv12.2 is heterologously expressed.
Cloning and Expression of KCNH3
Miyake et al. (1999) used the amino acid sequence of human KCNH2 to search the EST database and identified ESTs encoding KCNH3 and named it BEC1. Afterwards, they isolated a cDNA containing the entire KCNH3 coding sequence. The predicted 1083 amino acid KCNH3 protein contains six transmembrane domains, the pore region of a voltage-gated potassium channel, a CNB domain, and a putative N-glycosylation site. The amino acid sequence of KCNH3 shares 95% homology with rat KCNH3, 48% homology with human BEC2, 46% homology with rat Elk, 33% homology with human ERG, and the homology of rat Eag is 30%. The researchers believe that KCNH3, BEC2 and Elk are a subfamily of the eag family.
By Northern blot analysis of human tissues, the predominant KCNH3 transcript was detected in the brain and not expressed in other tissues. Inside the brain, KCNH3 is mainly expressed in cortical structures, such as the cerebral cortex, amygdala, and hippocampus, and striatal regions, including the putamen and caudate nuclei. Furthermore, KCNH3 expression was not detected in the spinal cord or in the corpus callosum, which mainly contains axons and glia. Therefore, Miyake et al. concluded that KCNH3 expression is largely confined to the telencephalon. In situ hybridization of rat brain slices showed that KCNH3 was significantly expressed in the hippocampus of rats, mainly in the hippocampal CA1, CA3 pyramidal cell layer and dentate gyrus granular cell layer. In the cerebral cortex, KCNH3 is widely expressed in layers II to VI, with specific expression in neuronal cell bodies with typical pyramidal shapes.
KCNH3 Structure and Function
The structural analysis of KCNH3 shows that the protein encoded by KCNH3 has a photo-oxygen voltage (LOV) and a cyclic nucleotide binding (CNB) domain at its N- and C-termini, respectively. The KCNH3-encoded protein has the longest S5-P loop of all known mammalian Kv channels with the most N-linked glycosylation sites (three sites). Studies have found that N-glycosylation affects the function of Kv12.2. Mainly because the removal of sugar chains resulted in a depolarizing transition of steady-state activation without a significant decrease in current amplitude.
KCNH3 and Disease
Existing studies suggest that human Kv12.2 may be associated with epilepsy.
Kv12.2 is a potent regulator of hippocampal pyramidal neuronal excitability. Genetic deletion and pharmacological blockade of Kv12.2 significantly reduced the firing threshold of these neurons. Kv12.2-/- mice exhibited persistent signs of neuronal hyperexcitability, including frequent interictal spikes, spontaneous seizures, and increased sensitivity to the chemoconvulsant pentylenetetrazole.
Clinical studies have shown that the typical symptom of FOXG1 syndrome is a congenital somatic malformation, a disease of abnormal brain structural function. Babies born with low birth weight and slower head development than normal babies have microcephaly in early infancy. This condition can be classified as a type of congenital abnormal brain function, that is, the corpus callosum, which connects the left and right brains in the body, is underdeveloped or too thin, the number of gyri in the cerebral cortex is reduced, and the number of white matter in the brain is also less than that of normal infants. It has been found that the transcription of KCNH3, the gene encoding Kv12.2, may be activated by the transcription factor FOXG1 in mature CNS neurons, suggesting a possible role in FOXG1 syndrome pathology.
References
- Gutman GA et al.; International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev.. 2005, 57:473-508.
- Zhang X et al.; Deletion of the potassium channel Kv12.2 causes hippocampal hyperexcitability and epilepsy. Nat. Neurosci.2010, 13:1056-8.
- Engeland B et al.; Cloning and functional expression of rat ether-à-go-go-like K+ channel genes. J. Physiol.. 1998, 513:647-54.
- Miyake, A., et al.; New ether-a-go-go K(+) channel family members localized in human telencephalon. J. Biol. Chem. 1999, 274: 25018-25025.
- Vezzali R, et al.; The FOXG1/FOXO/SMAD network balances proliferation and differentiation of cortical progenitors and activates Kcnh3 expression in mature neurons. Oncotarget. 2016, 7(25):37436-37455.
Inquiry