In the search for the mutational basis of familial neonatal benign epilepsy, the researchers discovered a novel voltage-gated potassium channel gene on human chromosome 20. The analysis found that the single cDNA isolated in this region by D20S24 probe has significant homology with KVLQT1 (also known as KCNQ1 and KCNA9). Subsequent studies found that KCNQ2 cDNA can hybridize with approximately 1.5, 3.8, and 9.5 kb transcripts on Northern blots made from the brain. By cloning the location of the 20q13.3 region, the KCNQ2 gene was independently isolated and found to be expressed in the brain. Yang et al. (1998) described the cloning, tissue distribution and functional expression of KCNQ2 and KCNQ3, both of which are related to benign neonatal epilepsy. The deduced KCNQ2 protein containing 871 amino acids has the characteristics of voltage-gated potassium channels. Northern blot analysis of 8 human tissues only detected 8.5 kb KCNQ2 transcripts in the brain. In the brain, KCNQ2 has the highest expression in the cerebellar cortex, amygdala, caudate nucleus and hippocampus.
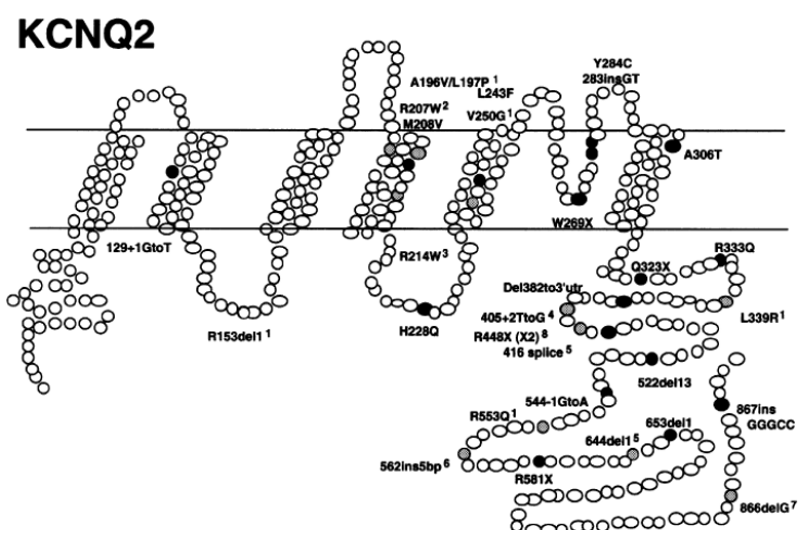
Figure 1. Location of 29 mutations in the KCNQ2 gene. (Singh NA, et al.; 2003)
The KCNQ2 gene belongs to a large family of genes that provide instructions for the generation of potassium channels. These channels transport positively charged potassium ions inside and outside the cell, and play a key role in the cell's ability to generate and transmit electrical signals.
The specific function of potassium channel depends on its protein composition and its location in the body. The channels formed by the KCNQ2 protein are active in nerve cells (neurons) in the brain, where they transport potassium ions out of the cell. These channels transmit a special type of electrical signal called m-current, which prevents neurons from continuing to send signals to other neurons. The m current ensures that neurons will not continue to be active or easily excitable.
KCNQ2 Channel Structure
Potassium ion channels are composed of several protein components (subunits). Each channel contains four alpha subunits, which form holes through which potassium ions pass. The four alpha subunits of the KCNQ2 gene can form a channel. However, the KCNQ2 α subunit can also interact with the α subunit produced by the KCNQ3 gene to form a functional potassium channel, which delivers stronger m currents.
KCNQ2 and Disease
Benign Familial Neonatal Seizures
In most patients with benign familial neonatal epilepsy (BFNS), mutations in the KCNQ2 gene have been confirmed. BFNS is a disease characterized by recurrent neonatal seizures (epilepsy). Seizures start around the 3rd day after birth and usually disappear within 1 to 4 months. In families with this disease, more than 60 KCNQ2 gene mutations have been found. Sometimes, the mutated protein cannot reach the cell surface to form a channel, or the channel may be located in the wrong part of the neuron, or the channel formed by the mutant protein may not work properly. These mutations cause the m current to decrease or change, leading to excessive neuron excitement. Researchers believe that reducing m current by 25% is enough to cause BFNS. When the neurons in the brain are abnormally excited, epilepsy occurs. It is unclear why the epilepsy stopped at 4 months of age. Studies have shown that potassium channels formed by KCNQ2 and KCNQ3 proteins play an important role in preventing neonatal neuronal overexcitation, but there are other mechanisms that prevent continuous neuronal activity in infancy.
References
- Biervert C, et al.; A potassium channel mutation in neonatal human epilepsy. Science. 1998, 279(5349):403-6.
- Castaldo P, et al.; Benign familial neonatal convulsions caused by altered gating of KCNQ2/KCNQ3 potassium channels. J Neurosci. 2002, 22(2):RC199.
- Chung HJ, et al.; Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc Natl Acad Sci USA. 2006,103(23):8870-5.
- Lerche H, et al.; A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann Neurol. 1999, 46(3):305-12.
- Millichap JJ, Cooper EC. KCNQ2 Potassium Channel Epileptic Encephalopathy Syndrome: Divorce of an Electro-Mechanical Couple? Epilepsy Curr. 2012, 12(4):150-2.
- Rogawski MA. KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci. 2000, 23(9):393-8.
- Saitsu H, et al.; Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol. 2012, 72(2):298-300.
- Schroeder BC, et al.; Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998, 396(6712):687-90.
- Yang, W.-P., et al.; Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J. Biol. Chem. 1998, 273: 19419-19423.
- Singh NA, et al.; KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003, 126(Pt 12):2726-37.
Inquiry