KCNQ5
| Catalog | Product Name | Gene Name | Species | Morphology | Price |
|---|---|---|---|---|---|
| ACC-RI0147 | Human KCNQ3/KCNQ5 Stable Cell Line-HEK293 | KCNQ5 | Human | Epithelial | INQUIRY |
Potassium channels are ubiquitous in all eukaryotic cells and have multiple functions, including maintaining cell osmotic pressure and volume, controlling cell membrane potential, and transmitting electrical signals in nerve cells. Potassium channels are the largest and most diverse group of ion channels, spanning the lipid bilayer of the cell membrane. Action potentials in nerve cells occur through polarization and repolarization processes, which are regulated by many different subtypes of potassium channels. Each channel subtype has its own kinetics, voltage dependence, and sensitivity to pharmacological blockers. More than 90 human genes encode potassium channel subunits, which have different expressions in different tissue types, and more than one-third of these genes are known to cause human diseases when they are mutated.
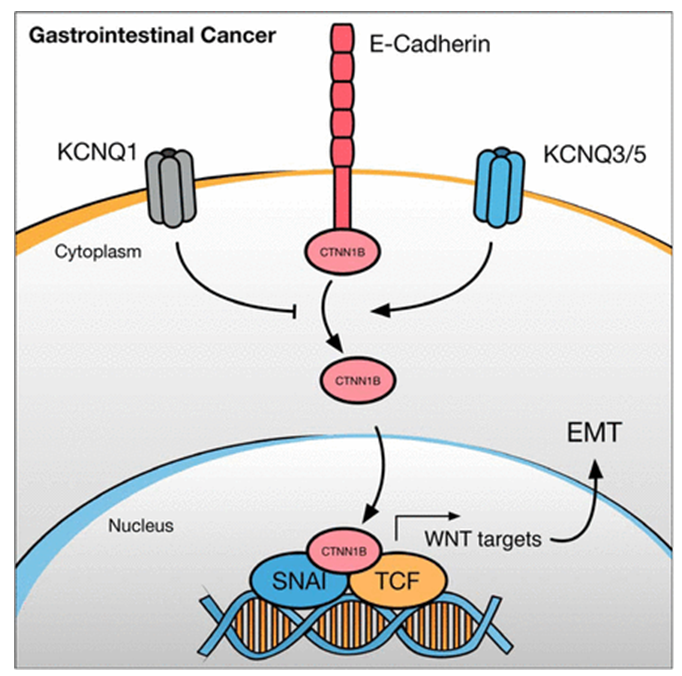
Figure 1. KCNQ gene family members act as both tumor suppressors and oncogenes in gastrointestinal cancers. (David Shorthouse., et al.; 2020)
The voltage-gated potassium channels Kv7.1–Kv7.5 are coded by KCNQ1-KCNQ5, respectively. The biallelic loss-of-function mutations in KCNQ1 can cause Jervell and Lange-Nielsen syndromes (sensory neurological hearing disorders with arrhythmia), while heterozygous mutations can cause a series of arrhythmias or alter insulin secretion. KCNQ2 and KCNQ3 form heterodimers, and heterozygous mutations in either gene can cause benign familial neonatal seizures or epileptic encephalopathy. There is evidence that the Kv7.5 subunit can be heterodimerized with the Kv7.3 subunit.
The KCNQ5 gene encodes a voltage-gated potassium channel, which is very important for the regulation of basic M-type current and discharge rate. M current is a slow activation and deactivation current, which plays a role in regulating the excitability of neurons by preventing repetitive action potential discharge during long-term depolarization input. Voltage-dependent potassium channels are key regulators of resting membrane potential and regulate the excitability of electrically active cells. These channels are usually tetrameric and can interact with auxiliary subunits to enhance or change the current mediated by the pore-forming subunits.
KCNQ5 Clone Expression
Lerche et al. used the homology with the KCNQ potassium channel family to identify candidate ESTs and screened the brain cDNA library to obtain the full-length KCNQ5 cDNA. The deduced protein molecular weight of 932 amino acids is about 102 kD. KCNQ5 contains 6 transmembrane domains, and there is a pore domain between the transmembrane segments S5 and S6. It has several potential sites for protein kinase C phosphorylation, but lacks the camp-dependent phosphorylation n-terminal sites present in KCNQ1 and KCNQ2. The amino acid homology between KCNQ5 and KCNQ4 is 65%, the amino acid homology with KCNQ3 and KCNQ2 is 50%, and the amino acid homology with KCNQ1 is 40%, with the greatest homology in the transmembrane and pore regions. Northern blot analysis revealed a 7.5 kb transcript in adult skeletal muscle and brain. In the brain, KCNQ5 is most strongly expressed in the cerebral cortex, occipital pole, frontal lobe and temporal lobe. Compared with KCNQ2, KCNQ5 expression is reduced or absent in the cerebellum.
After that, the researchers cloned KCNQ5 from the thalamus cDNA library, and cloned it in the brain cDNA library through 5-prime and 3-prime RACE. They found several splice variants with different cytoplasmic c-terminal tails. One variant is isolated from the brain, while the other is isolated from the skeletal muscle. In situ hybridization of rat brain slices confirmed that Kcnq5 is widely expressed in the brain and upper cervical ganglia. There is also a weak signal in the rat cerebellum, which is not expressed in the cerebellum compared with Northern blot analysis of human brain regions.
KCNQ5 and Disease
Mutations in KCNQ5 (encoding Kv7.5) can cause congenital neurological dysfunction, intellectual disability, or epileptic encephalopathy. KCNQ's other homologous genes KCNQ2 and less common mutations of KCNQ3 can also cause epilepsy through loss of function or gain of function. In neurons, Kv7.5 is very important for the regulation of m-type currents, so the firing rate is generated. m current is a slowly activated and inactivated neuronal potassium current. It plays a vital role in regulating neuronal excitability by preventing repetitive action potential discharge during long-term depolarization input. Inhibition of m current leads to increased neuronal excitability and is associated with a wide range of early epileptic diseases, ranging from benign family neonatal convulsions to severe epileptic encephalopathy and/or intellectual disability in mice. Kcnq5 negative mutations have been shown to alter the hippocampus area synaptic inhibition and excitability.
References
- Anna Lehman, et al.; Loss-of-Function and Gain-of-Function Mutations in KCNQ5 Cause Intellectual Disability or Epileptic Encephalopathy. Am J Hum Genet. 2017, 101(1): 65-74.
- Lehman, A., et al.; Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am. J. Hum. Genet. 2017, 101: 65-74.
- Lerche, C., et al.; Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 2000, 275: 22395-22400.
- Schroeder, B. C., et al.; KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000, 275: 24089-24095.
- Zhang, H.,et al.; PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003, 37: 963-975.
- David Shorthouse., et al.; KCNQ gene family members act as both tumor suppressors and oncogenes in gastrointestinal cancers. 2020, DOI:10.1101/2020.03.10.984039.
Inquiry