The SCN8A gene belongs to a family of genes that provide instructions to make sodium channels. These sodium channels allow positively charged sodium ions to enter the cell, allowing them to play a key role in the process of cell generation and transmission of electrical signals. Among them, the SCN8A gene provides instructions to make a part of the sodium channel (α subunit) called Nav1.6. This subunit forms pores in the cell membrane through which sodium ions flow. Clinical studies have found that the Nav1.6 channel mainly exists in the nerve cells (neurons) of the brain and spinal cord (central nervous system), connects the neurons of the central nervous system and muscles, and detects the sensory cells of sense of touch, pain, heat and sound. The Nav1.6 channel controls the flow of sodium ions into the cell, which makes it possible for neurons to communicate by generating and transmitting electrical signals.
SCN8A Cloning and Expression
The researchers first isolated and sequenced a genomic fragment containing the human voltage-gated sodium channel SCN8A exon. Through sequencing and sequence comparison studies, it is found that SCN8A encodes a protein composed of 1980 amino acids, and the sequence similarity between the protein and the mouse protein is close to 99%. It contains several consistent sites for phosphorylation of serine and threonine residues, which are also conserved among other sodium channel family members. Later, two alternative splicing exons of SCN8A, 18N and 18A were also identified, which encode the transmembrane fragments S3 and S4 of domain III. Among them, exon 18N is expressed in fetal brain and non-neuronal tissues. The transcript of exon 18N has a conserved in-frame stop codon that predicts the synthesis of a truncated 2-domain protein. Between embryonic day (E) 12.5 and E14.5, the proportion of transcripts containing exon 18N in the mouse fetal brain is the highest; at E18.5 and later, 18A transcripts dominate.
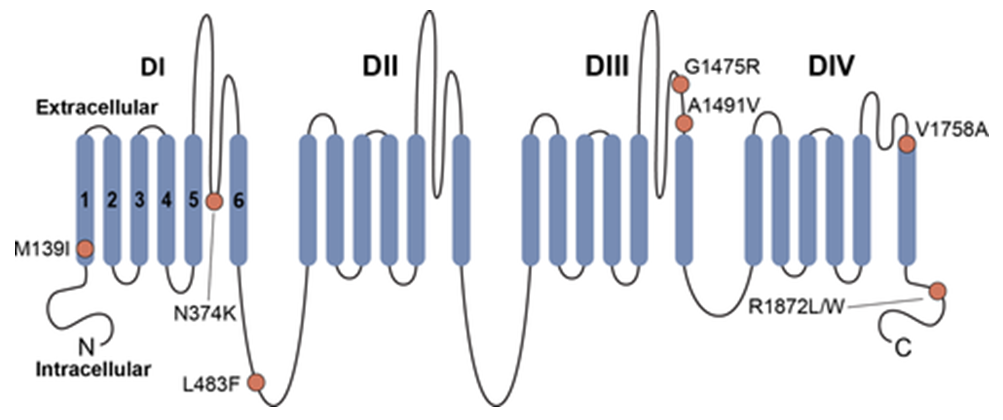
Figure 1. Schematic of Nav1.6. (Zaman T, et al.; 2019)
SCN8A and Disease
SCN8A-related Epilepsy with Encephalopathy
More than 100 mutations in the SCN8A gene that cause SCN8A-related epilepsy and encephalopathy have been discovered. This disease is characterized by recurrent seizures (epilepsy), abnormal brain function (encephalopathy), and intellectual disability. The signs and symptoms of this condition usually begin in infancy. Most of these mutations in the SCN8A gene change the amino acid of a single protein in the Nav1.6 channel. Mutations in SCN8A-related epilepsy lead to changes in channels that are open for longer than usual, thereby increasing the flow of sodium ions into neurons. Continuously open channels will abnormally increase electrical signals, leading to excessive activation (excitement) of brain neurons. The increase in neural activity leads to seizures in SCN8A-related epilepsy patients with encephalopathy. It is not clear how mutations in the SCN8A gene cause intellectual disability, movement problems, and other features of SCN8A-related epilepsy with encephalopathy. Since some affected children experience a loss of previously acquired skills (regression in development) after a seizure, some people think that seizures may impair brain function, but it is not clear whether this is the case.
Other Disorders
Studies have found that mutations in the SCN8A gene can cause intellectual disability and exercise problems in some people. Different from the mutation that causes SCN8A-related encephalopathy epilepsy, the SCN8A gene mutation that causes this situation causes the activity of the Nav1.6 sodium channel to be lower than the normal level, and the amount of sodium flowing into the neuron is reduced. This change does not increase neuronal signals, so individuals with these mutations will not develop epilepsy. Researchers suspect that the reduction of some neuronal signals in the brain that coordinate movement may be the cause of movement problems. Other mutations in the SCN8A gene can cause movement disorders and seizures called benign infantile epilepsy. This disease usually appears in the first year of life and stops by the age of 3. Unlike most other individuals with mutations in the SCN8A gene, these individuals have normal intellectual functions. It is unclear why some SCN8A gene mutations cause these milder signs and symptoms, while other mutations cause more severe features of SCN8A-related epilepsy and encephalopathy.
References
- Butler KM, et al.; De novo and inherited SCN8A epilepsy mutations detected by gene panel analysis. Epilepsy Res. 2017, 129:17-25.
- O'Brien JE, et al.; Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. 2013, 4:213.
- Ohba C, et al.; Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014, 55(7):994-1000.
- Wagnon JL, et al.; Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 2015, 3(2):114-23.
- Zaman T, et al.; A single-center SCN8A-related epilepsy cohort: clinical, genetic, and physiologic characterization. Ann Clin Transl Neurol. 2019, 6(8):1445-1455.
Inquiry