KCNQ4
| Catalog | Product Name | Gene Name | Species | Morphology | Price |
|---|---|---|---|---|---|
| ACC-RI0045 | Human KCNQ4 Stable Cell Line-HEK293 | KCNQ4 | Human | Epithelial | INQUIRY |
| ACC-RI0146 | Human KCNQ2/KCNQ4 Stable Cell Line-CHO | KCNQ4 | Human | Epithelial-like | INQUIRY |
| ACC-RI0148 | Human KCNQ4 Stable Cell Line-CHO | KCNQ4 | Human | Epithelial-like | INQUIRY |
KCNQ4 is a member of the voltage-gated potassium channel gene family and provides coding instructions for making proteins that are part of the potassium channel family. Potassium ion channels transport positively charged potassium ions between adjacent cells. Studies have found that these channels play a key role in the ability of cells to generate and transmit electrical signals. The specific function of a potassium ion channel depends on its protein composition and its location in the body. The potassium channel composed of KCNQ4 protein mainly exists in certain cells of the inner ear, and along part of the nerve pathway (auditory pathway) from the ear to the brain. In addition, KCNQ4 potassium channels are also less distributed in the heart and some other muscles. Because KCNQ4 potassium channels exist in the inner ear and auditory pathways, researchers have been paying attention to their role in hearing. Hearing needs to convert sound waves into neuroelectric signals, which are then transmitted to the brain. This conversion involves many processes, including maintaining proper levels of potassium ions in the inner ear. The KCNQ4 channel helps maintain these levels and plays a key role in the effective transmission of nerve electrical signals from the inner ear to the brain.
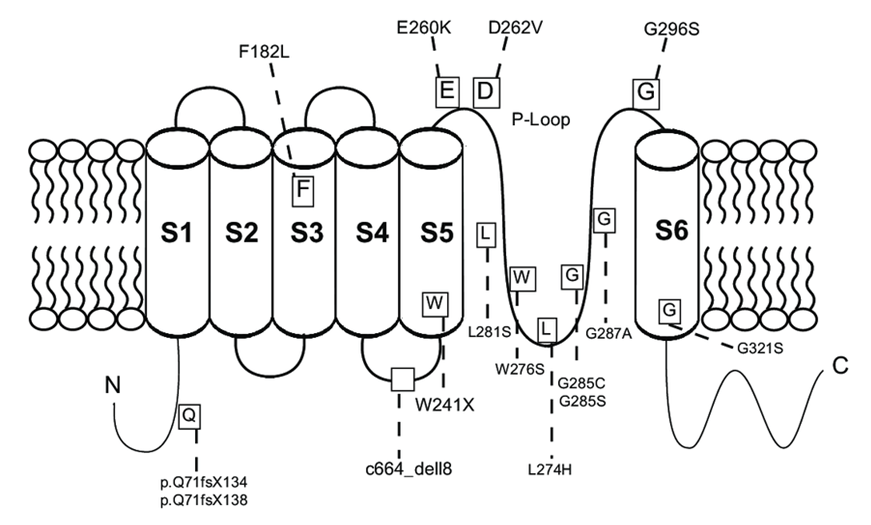
Figure 1. Structure of KCNQ4. (Laura M Dominguez, et al.; 2012)
Kubisch et al. cloned KCNQ4 cDNA, which encodes a 695 amino acid polypeptide with an estimated mass of 77 kD. Its amino acid homology with KCNQ1, KCNQ2 and KCNQ3 are 38%, 44% and 37%, respectively. Through structural analysis, it is speculated that KCNQ4 has 6 transmembrane domains, and there is a P loop between the transmembrane domains S5 and S6. In the potassium ion channel, four highly conserved P-rings join together to form an ion-selective pore. They are tetramers composed of the same or homologous subunits. Like other KCNQ channels, KCNQ4 has a long-term predicted cytoplasmic C-terminus, which accounts for about half of the protein.
KCNQ4 Function
In the mouse cochlea, the KCNQ4 transcript is only present in outer hair cells. Using specific antibodies, Kharkovets et al. found that Kcnq4 is located on the basement membrane of these sensory cells. In the vestibular organs, Kcnq4 is confined to type I hair cells, and the afferent goblet nerve endings wrap these sensory cells. Kcnq4 is also expressed in many (but not all) neurons in the nucleus of the central auditory pathway, and is absent in most other brain regions. For example, it exists in the cochlear nucleus, lateral colliculus nucleus and inferior colliculus. The researchers pointed out that this is the first ion channel specifically expressed in the sensory pathway. In addition, the researchers recorded channel currents produced by crna-injected Xenopus oocytes and found that phosphatidylinositol (4,5)-diphosphate activated all KCNQ channel family members analyzed, including KCNQ4.
KCNQ4 and Disease
Nonsyndromic Hearing Loss
Some KCNQ4 gene mutations have been reported in individuals with non-syndromic hearing loss. Non-syndromic hearing loss refers to hearing loss that is not related to other signs and symptoms. Mutations in this gene can cause a non-syndromic hearing loss called DFNA2. This form of hearing loss usually starts after the child learns to speak (post-language), and it especially affects the ability to hear high-frequency sounds. DFNA2 is described as progressive, which means that it will become more severe over time. Most KCNQ4 gene mutations change the components used to make KCNQ4 protein. Some mutations prevent this channel from reaching the cell membrane because it needs to transport potassium ions on the cell membrane. Other mutations lead to the formation of abnormal channels that cannot transport these ions efficiently. The loss of functional KCNQ4 channels seems to cause the accumulation of potassium ions in certain cells of the inner ear, thereby damaging these cells, leading to gradual hearing loss in DFNA2 patients.
In addition, clinical studies have also found that KCNQ4 gene mutations are also associated with Age-related hearing loss.
References
- Coucke PJ, et al.; Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet. 1999, 8(7):1321-8.
- Gao Y, et al.; Impaired surface expression and conductance of the KCNQ4 channel lead to sensorineural hearing loss. J Cell Mol Med. 2013 Jul;17(7):889-900.
- Kim HJ, et al.; Cellular and molecular mechanisms of autosomal dominant form of progressive hearing loss, DFNA2. J Biol Chem. 2011, 286(2):1517-27.
- Kubisch C, et al.; KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999, 96(3):437-46.
- Kharkovets, T., et al.; KCNQ4, a K(+) channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc. Nat. Acad. Sci. 2000, 97: 4333-4338.
- Laura M Dominguez, et al.; Genetics of hearing loss: Focus on DFNA2. The Application of Clinical Genetics. 2012, 5:97-104.
Inquiry