KCNH2 gene belongs to a large family of genes that provide for the synthesis of potassium channel proteins. These channels are responsible for transporting positively charged potassium ions out of the cell in the body, and play a key role in the cell's ability to generate and transmit electrical signals. Studies have found that the specific function of a potassium channel depends on its protein composition and its location in the body. The channel composed of the KCNH2 protein (also called hERG1) is active in the heart (myocardium). They participate in recharging the heart muscle after each heartbeat to maintain a regular heart rate. KCNH2 protein is also produced in nerve cells in the brain and spinal cord (central nervous system) and certain immune cells (microglia). The protein produced by the KCNH2 gene and another gene KCNE2 interacts to form a functional potassium channel. The four alpha subunits produced by the KCNH2 gene constitute the structure of each channel. A β subunit produced by the KCNE2 gene attaches to the channel and regulates its activity.
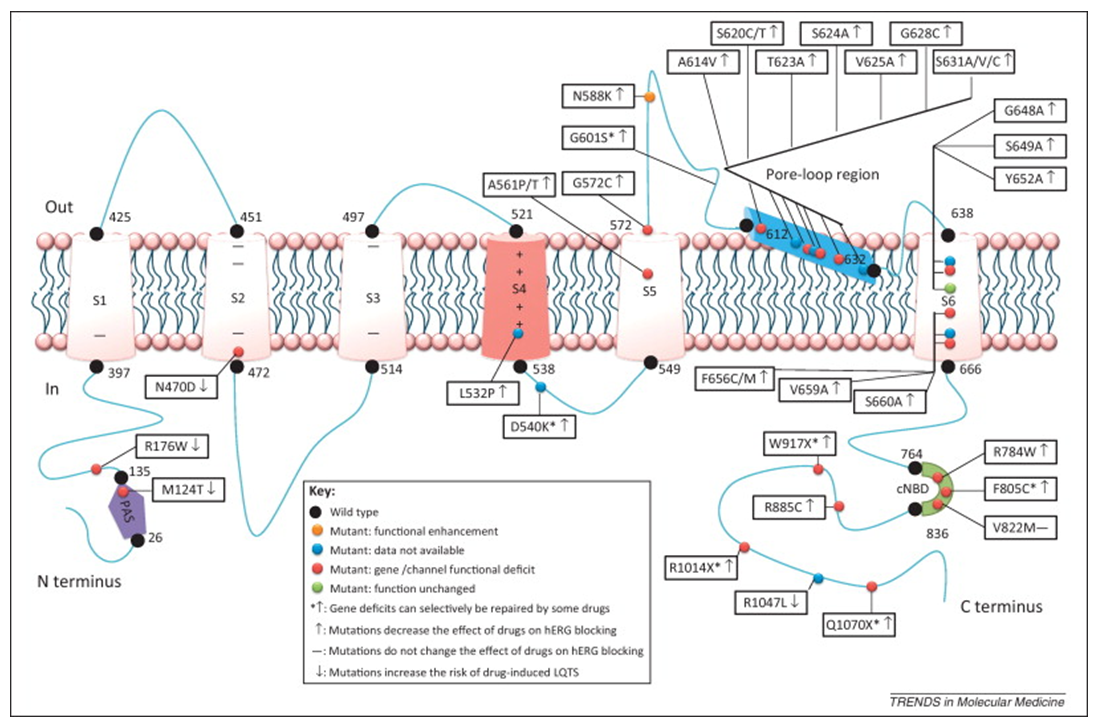
Figure 1. Current pharmacogenomic studies on hERG potassium channels. (Fa-Zhong He, et al.; 2013)
KCNH2 Cloning and Expression
Itoh et al. described the genome structure of HERG for the first time. They found that the gene contains 15 exons on chromosome 7q35 and is about 19 kb in length. After that, the researchers analyzed the genome sequence and determined that the HERG gene contains 16 exons (including one candidate exon 1b), ranging from 100 bp to 553 bp. Warmke and Ganetzky identified a new human cDNA from the hippocampal cDNA library by homology with the Drosophila'ethera -go-go' (eag) gene. After the author called this gene cDNA HERG. Scientists discovered a brain-specific subtype of KCNH2, called KCNH2-3.1. The new subtype originates from the upstream of exon 3 of the known subtype KCNH2-1A and contains all downstream exons of the full-length gene. Through exon 15, the prediction of the longest open reading frame of KCNH2-3.1 indicated that most of the 5-prime extension of exon 3 was untranslated, and the first methionine and the conserved part of KCNH2-1A were in the frame. Therefore, it is speculated that KCNH2-3.1 deleted the first 102 amino acids of KCNH2-1A and replaced it with 6 unique amino acids. Western blot analysis of human hippocampal and frontal cortex cells confirmed the predicted difference in protein size. The KCNH2-3.1 subtype is preferentially expressed in the human brain. It was not detected in the brains of mice, but was found in large numbers in the brains of rhesus monkeys, indicating that it is unique to primates. In various human brain tissues, the KCNH2-3.1 transcription level was significantly higher than that of adults before delivery, but it decreased and stabilized shortly after birth. In contrast, the expression of KCNH2-1A continues to increase throughout prenatal development until it reaches a maximum level and continues after birth. Research results show that KCNH2-3.1 plays a specific role in the early stages of human brain development.
KCNH2 and Disease
Mutations in the KCNH2 gene can cause Romano-Ward syndrome, which is the most common heart disease called long QT syndrome. Mutations in this gene account for approximately 25% of Romano-Ward syndrome cases. In this case, the heart muscle takes longer than usual to recharge, which may cause arrhythmia. More than 900 mutations in the KCNH2 gene that cause Romano-Ward syndrome have been discovered. Some of these mutations change a single amino acid sequence in the KCNH2 protein, while others delete some amino acids in the protein. These changes prevent proteins from assembling into ion channels or change the structure or function of the channels. Therefore, these channels cannot properly regulate the flow of potassium ions in cardiomyocytes. Reduced ion transmission can change the transmission of electrical signals in the heart, increasing the risk of arrhythmia, which can lead to syncope (syncope) or sudden death.
Mutations in the KCNH2 gene can also cause a heart disease called short QT syndrome. In this case, the charging time of the myocardium between two heartbeats is shorter than usual. This change increases the risk of abnormal heart rhythms, which can lead to syncope or sudden death. In the few affected families, at least 8 mutations in the KCNH2 gene cause short QT syndrome. These mutations changed a single amino acid in the KCNH2 protein. Genetic mutantion change the function of the ion channel formed by the KCNH2 protein and increase the channel's activity. Therefore, at the critical moment of the heartbeat, more potassium ions will flow out of the cardiomyocytes, resulting in arrhythmia.
References
- Brugada R, et al.; Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004, 109(1):30-5.
- Cordeiro JM, et al.; Modulation of I(Kr) inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc Res. 2005, 67(3):498-509.
- Hong K, et al.; Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005, 16(4):394-6.
- McBride CM, et al.; Mechanistic basis for type 2 long QT syndrome caused by KCNH2 mutations that disrupt conserved arginine residues in the voltage sensor. J Membr Biol. 2013, 246(5):355-64.
- Paulussen AD, et al.; Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J Mol Med (Berl). 2004, 82(3):182-8.
- Paulussen AD, et al.; HERG mutation predicts short QT based on channel kinetics but causes long QT by heterotetrameric trafficking deficiency. Cardiovasc Res. 2005, 67(3):467-75.
- Itoh, T., et al.; Genomic organization and mutational analysis of HERG, a gene responsible for familial long QT syndrome. Hum. Genet. 1998, 102: 435-439.
- Itzhaki, I., et al.; Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011, 471: 225-229.
- Fa-Zhong He, et al.; Current pharmacogenomic studies on hERG potassium channels. Trends in Molecular Medicine. 2013, 19(4): P227-238.
Inquiry