Inwardly rectifying potassium channels (Kir) are widely expressed in several excitable and non-excitable tissues, and play a key role in maintaining the resting membrane potential and thus regulating cell excitability. Based on sequence similarity and functional characteristics, approximately 15 Kir clones were identified and divided into seven different families: Kir1.x-Kir7.x. The Kir2.x family includes five isoforms, which have a highly voltage-dependent blocking of intracellular polyamines and Mg2+ on the channel pores, thus having strong biophysical characteristics of inward rectification.
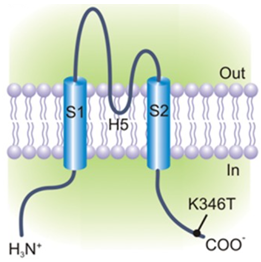
Figure 1. Schematic representation of the membrane topology of a human Kir2.1 subunit. (Ambrosini E, et al.; 2014)
The KCNJ2 gene is a gene family that directs the formation of the inward rectifier potassium channel Kir2.1. These channels can transport positively charged potassium ions out of the cell, a process that plays an important role in the cell's ability to generate and transmit electrical signals. Studies have found that the specific function of a potassium ion channel depends on its protein composition and its location in the body. The channel formed by the KCNJ2 protein is usually active in the muscles (skeletal muscle) and heart muscle (heart muscle) used for exercise. In skeletal muscles, these channels play an important role in the pattern of muscle tension (contraction) and relaxation, allowing the body to move. In the heart, these channels charge the heart muscle after each heartbeat to maintain a regular rhythm. The channel formed with KCNJ2 protein may also be involved in bone development, but their role in this process is unclear. Researchers have determined that a molecule called PIP2 must bind to the channel formed by the KCNJ2 protein in order for the channel to work properly. PIP2 activates the ion channel and helps it stay open, allowing ions to flow through the cell membrane.
KCNJ2 and Disease
Andersen-Tawil Syndrome
Clinical studies have found that more than 60 mutations in the KCNJ2 gene can cause Anderson-Tavel syndrome. This disease is a disease characterized by muscle weakness (periodic paralysis), arrhythmia, and physiological abnormalities affecting the face, other parts of the head, and limbs. Most mutations change a protein component of the KCNJ2 protein, which will lead to the production of non-functional potassium channels. Some mutations change the shape of the channel, it cannot transport potassium ions, while other mutations prevent the channel from being inserted correctly into the cell membrane. In addition, there are many KCNJ2 mutations that prevent PIP2 from effectively binding and activating potassium channels. If the KCNJ2 protein cannot bind to PIP2, the channel will remain closed and potassium ions will not flow through the cell membrane. Researchers believe that the problem of PIP2 binding is the main cause of Anderson-Tavel syndrome. The loss of potassium channel function in skeletal muscle and cardiomyocytes interferes with the normal flow of potassium ions out of these cells, leading to periodic paralysis and arrhythmia. It is not clear how mutations in the KCNJ2 gene cause the common physiological abnormalities in Anderson-Tavel syndrome.
Short QT Syndrome
Mutations in the KCNJ2 gene can also cause a heart disease called short QT syndrome, which is a type of arrhythmia. In this case, the recovery time of the myocardium between beats is shorter than normal. This change increases the risk of abnormal heart rhythms, which can lead to fainting or sudden death. In a small number of affected families, at least two mutations in the KCNJ2 gene have been found to cause short QT syndrome. These mutations change a single amino acid in the KCNJ2 protein, thereby increasing the activity of the channel composed of the protein. As a result, more potassium ions will flow out of the cardiomyocytes at the critical moment of the heartbeat, leading to arrhythmia.
References
- Bendahhou S, et al.; Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J Biol Chem. 2003, 278(51): 51779-85.
- Casini S, Postma AV. Decreased inward rectification of Kir2.1 channels is a novel mechanism underlying the short QT syndrome. Cardiovasc Res. 2012, 93(4):535-6.
- Chun TU, et al.; Polymorphic ventricular tachycardia and KCNJ2 mutations. Heart Rhythm. 2004, 1(2):235-41.
- Donaldson MR, et al.; PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology. 2003, 60(11):1811-6.
- Hattori T, et al.; A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. 2012, 93(4):666-73.
- Ambrosini E, et al.; Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum Mol Genet. 2014, 23(18):4875-86.
Inquiry